Variational Quantum Eigensolver (VQE)
สำหรับโมดูลนี้ นักเรียนต้องมี Python environment ที่ใช้งานได้ และติดตั้งแพ็กเกจเวอร์ชันล่าสุดต่อไปนี้:
qiskitqiskit_ibm_runtimeqiskit-aerqiskit.visualizationnumpypylatexenc
สำหรับการติดตั้งแพ็กเกจเหล่านี้ ดูได้ที่คู่มือ ติดตั้ง Qiskit หากต้องการรัน jobs บนคอมพิวเตอร์ควอนตัมจริง นักเรียนจะต้องตั้งค่าบัญชี IBM Cloud ตามขั้นตอนในคู่มือ ตั้งค่าบัญชี IBM Cloud ของคุณ
โมดูลนี้ผ่านการทดสอบแล้ว และใช้เวลา QPU ประมาณ 8 นาที ค่านี้เป็นการประมาณ และการใช้งานจริงของคุณอาจแตกต่างกัน
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-aer qiskit-ibm-runtime scipy
# Uncomment and modify this line as needed to install dependencies
#!pip install 'qiskit>=2.1.0' 'qiskit-ibm-runtime>=0.40.1' 'qiskit-aer>=0.17.0' 'numpy' 'pylatexenc'
บทนำ
นับตั้งแต่การพัฒนาแบบจำลองทางกลควอนตัมในช่วงต้นศตวรรษที่ 20 นักวิทยาศาสตร์เข้าใจแล้วว่าอิเล็กตรอนไม่ได้โคจรรอบนิวเคลียสของอะตอมตามเส้นทางที่แน่นอน แต่กลับอยู่ในบริเวณความน่าจะเป็นที่เรียกว่าออร์บิทัล ออร์บิทัลเหล่านี้สอดคล้องกับระดับพลังงานที่ไม่ต่อเนื่องและเฉพาะเจาะจง ซึ่งอิเล็กตรอนสามารถครอบครองได้ อิเล็กตรอนจะอยู่ในระดับพลังงานต่ำสุดที่มีอยู่โดยธรรมชาติ ซึ่งเรียกว่า ground state อย่างไรก็ตาม หากอิเล็กตรอนดูดซับพลังงานเพียงพอ มันสามารถกระโดดขึ้นไประดับพลังงานที่สูงขึ้น เข้าสู่ excited state ซึ่งเป็นสถานะชั่วคราว และอิเล็กตรอนจะกลับสู่ระดับพลังงานที่ต่ำกว่าในที่สุด โดยปล่อยพลังงานที่ดูดซับออกมา มักอยู่ในรูปของแสง กระบวนการพื้นฐานของการดูดซับและการปล่อยพลังงานนี้มีความสำคัญต่อการทำความเข้าใจว่าอะตอมมีปฏิสัมพันธ์และสร้างพันธะอย่างไร
เมื่ออะตอมมารวมกันเป็นโมเลกุล ออร์บิทัลอะตอมมิกของพวกมันจะรวมกันเป็นออร์บิทัลโมเลกุล การจัดเรียงและระดับพลังงานของอิเล็กตรอนภายในออร์บิทัลโมเลกุลเหล่านี้กำหนดคุณสมบัติของโมเลกุลที่เกิดขึ้นและความแข็งแรงของพันธะเคมี ตัวอย่างเช่น ในการก่อตัวของโมเลกุลไฮโดรเจน () จากอะตอมไฮโดรเจนสองอะตอม อิเล็กตรอนจากแต่ละอะตอมครอบครองออร์บิทัลอะตอมมิก เมื่ออะตอมเข้าใกล้กัน ออร์บิทัลอะตอมมิกเหล่านี้จะทับซ้อนและรวมกันเป็นออร์บิทัลโมเลกุลใหม่ — ออร์บิทัลหนึ่งมีพลังงานต่ำกว่า (bonding orbital) และอีกออร์บิทัลหนึ่งมีพลังงานสูงกว่า (anti-bonding orbital) อิเล็กตรอนสองตัว หนึ่งตัวจากแต่ละอะตอมไฮโดรเจน จะครอบครอง bonding orbital ที่มีพลังงานต่ำกว่าเป็นหลัก ทำให้เกิดพันธะโควาเลนต์ที่เสถียรซึ่งยึดโมเลกุล ไว้ด้วยกัน ความแตกต่างของพลังงานระหว่างอะตอมที่แยกออกจากกันและโมเลกุลที่ก่อตัวขึ้น โดยเฉพาะพลังงานของอิเล็กตรอนในออร์บิทัลโมเลกุล กำหนดความเสถียรและคุณสมบัติของพันธะ
ในส่วนต่อไปนี้ เราจะสำรวจกระบวนการก่อตัวของโมเลกุลนี้ โดยมุ่งเน้นที่โมเลกุล เราจะใช้คอมพิวเตอร์ควอนตัมจริง ร่วมกับเทคนิคการปรับค่าแบบคลาสสิก เพื่อหาพลังงานของกระบวนการพื้นฐานแต่สำคัญนี้ การทดลองนี้จะแสดงให้เห็นอย่างเป็นรูปธรรมว่าการคำนวณเชิงควอนตัมสามารถนำไปประยุกต์ใช้แก้ปัญหาทางเคมีเชิงคำนวณได้อย่างไร
VQE - อัลกอริทึมควอนตัมแบบ variational สำหรับปัญหา eigenvalue
เทคนิคการประมาณสำหรับเคมี — หลักการ variational และ basis set
การมีส่วนร่วมของ Erwin Schrödinger ต่อกลศาสตร์ควอนตัมไม่ได้จำกัดอยู่แค่การแนะนำแบบจำลองอิเล็กตรอนใหม่เท่านั้น แต่โดยพื้นฐานแล้ว เขาได้ก่อตั้งกลศาสตร์คลื่นโดยพัฒนาสมการ Schrödinger แบบขึ้นกับเวลาที่มีชื่อเสียง:
ที่นี่ คือตัวดำเนินการ Hamiltonian ซึ่งแทนพลังงานรวมของระบบ และ คือฟังก์ชันคลื่นที่บรรจุข้อมูลทั้งหมดเกี่ยวกับสถานะควอนตัมของระบบ (หมายเหตุ: คืออนุพันธ์รวมตามเวลา และเราไม่ได้ระบุ energy eigenvalue ไว้อย่างชัดเจนที่นี่)
อย่างไรก็ตาม ในการประยุกต์ใช้งานจริงหลายกรณี เช่น การหาระดับพลังงานที่อนุญาตของอะตอมและโมเลกุล เราจะใช้สมการ Schrödinger แบบไม่ขึ้นกับเวลา (สมการ eigenvalue ของพลังงาน) แทน ซึ่งได้มาจากรูปแบบที่ขึ้นกับเวลาโดยการสมมติ stationary state Stationary state คือสถานะควอนตัมที่ความหนาแน่นความน่าจะเป็นในการพบอนุภาค ณ จุดใดจุดหนึ่งในอวกาศไม่เปลี่ยนแปลงตามเวลา
ในรูปแบบนี้ แทน energy eigenvalue ที่สอดคล้องกับสถานะควอนตัม Hamiltonian ประกอบด้วยการมีส่วนร่วมของพลังงานต่างๆ เช่น พลังงานจลน์ของอิเล็กตรอนและนิวเคลียส แรงดึงดูดระหว่างอิเล็กตรอนและนิวเคลียส และแรงผลักระหว่างอิเล็กตรอน
การแก้สมการ eigenvalue ของพลังงานช่วยให้เราคำนวณระดับพลังงานเชิงควอนตัมของระบบอะตอมและโมเลกุลได้ อย่างไรก็ตาม สำหรับโมเลกุล การแก้อย่างแม่นยำนั้นทำได้ยาก เนื่องจากฟังก์ชันคลื่น ที่อธิบายการกระจายเชิงพื้นที่ของอิเล็กตรอนมีความซับซ้อนและมีหลายมิติ
ด้วยเหตุนี้ นักวิทยาศาสตร์จึงใช้เทคนิคการประมาณเพื่อให้ได้ผลลัพธ์ที่ใช้งานได้จริงและแม่นยำ ในงานนี้ เราจะมุ่งเน้นที่วิธีการสำคัญสองวิธี:
-
หลักการ variational
วิธีนี้ประมาณฟังก์ชันคลื่นและปรับให้ใกล้เคียงกับพลังงานเป้าหมายมากที่สุดเท่าที่จะทำได้ ซึ่งปกติแล้วคือ ground state energy ของระบบ แนวคิดหลักเบื้องหลังหลักการ variational นั้นเรียบง่าย:
- ถ้าเราเดาฟังก์ชันคลื่น (หรือ "trial function") พลังงานที่คำนวณได้จากมันจะเท่ากับหรือสูงกว่า ground state energy () ของระบบเสมอ
- ด้วยการปรับพารามิเตอร์ ใน trial function, เราสามารถได้การประมาณ ground state energy ที่ดีขึ้นเรื่อยๆ
- ความแม่นยำขึ้นอยู่กับการเลือก trial wave function อย่างมาก trial function ที่เลือกไม่ดีอาจนำไปสู่การประมาณพลังงานที่ห่างไกลจากความเป็นจริง
-
การประมาณด้วย basis set
วิธีการประมาณที่สองมาในขั้นตอนของการสร้างฟังก์ชันคลื่น — แนวทาง basis set ในเคมีควอนตัม การแก้สมการ Schrödinger อย่างแม่นยำสำหรับโมเลกุลนั้นแทบเป็นไปไม่ได้ แทนที่จะทำเช่นนั้น เราประมาณฟังก์ชันคลื่นแบบหลายอิเล็กตรอนที่ซับซ้อน โดยสร้างมันขึ้นจากฟังก์ชันทางคณิตศาสตร์ที่ง่ายกว่าและกำหนดไว้ล่วงหน้า Basis set โดยพื้นฐานแล้วคือชุดของฟังก์ชันทางคณิตศาสตร์ที่รู้จักเหล่านี้ โดยทั่วไปมีศูนย์กลางอยู่บนอะตอมในโมเลกุล ซึ่งใช้เป็นส่วนประกอบพื้นฐานเพื่อแทนรูปร่างและพฤติกรรมของอิเล็กตรอนในระบบ ลองนึกภาพว่าคุณพยายามสร้างประติมากรรมที่ละเอียดโดยใช้เพียงชุดตัวต่อ LEGO มาตรฐาน ยิ่งคุณมีประเภทและขนาดของตัวต่อมากเท่าไหร่ (basis set ที่ใหญ่กว่า) คุณก็ยิ่งสามารถประมาณรูปร่างต้นฉบับได้แม่นยำมากขึ้น
ฟังก์ชัน basis เหล่านี้มักได้รับแรงบันดาลใจจากวิธีแก้เชิงวิเคราะห์สำหรับระบบง่ายๆ อย่างอะตอมไฮโดรเจน โดยอยู่ในรูปแบบเช่นฟังก์ชัน Gaussian หรือ Slater-type แม้ว่าจะยังคงเป็นการประมาณอยู่ก็ตาม แทนที่จะทำงานกับออร์บิทัลโมเลกุลแบบเต็มที่ "แม่นยำ" ในทางทฤษฎีแต่จัดการได้ยาก เราแสดงพวกมันเป็น linear combination (ผลรวมพร้อมสัมประสิทธิ์) ของฟังก์ชัน basis เหล่านี้ วิธีนี้เรียกว่าแนวทาง Linear Combination of Atomic Orbitals (LCAO) เมื่อฟังก์ชัน basis มีลักษณะคล้ายออร์บิทัลอะตอมมิก ด้วยการปรับค่าสัมประสิทธิ์ใน linear combination นี้ เราสามารถหาฟังก์ชันคลื่นและพลังงานประมาณที่ดีที่สุดได้ภายในข้อจำกัดของ basis set ที่เลือก
- ยิ่งใส่ฟังก์ชันมากใน basis set มากเท่าใด การประมาณก็ยิ่งดีขึ้น แต่นั่นก็มีต้นทุนในด้านความพยายามในการคำนวณที่สูงขึ้น
- Basis set ขนาดเล็กให้ค่าประมาณคร่าวๆ ในขณะที่ basis set ขนาดใหญ่ให้ผลลัพธ์ที่แม่นยำกว่าแต่ต้องการทรัพยากรการคำนวณมากขึ้น
สรุปได้ว่า เพื่อให้การคำนวณทำได้จริงและลดต้นทุนการคำนวณ เราใช้หลักการ variational ด้วยการประมาณฟังก์ชันคลื่น ซึ่งช่วยลดความซับซ้อนในการคำนวณและเอื้อให้มีการปรับค่าแบบวนซ้ำเพื่อลดพลังงาน ขณะเดียวกัน แนวทาง basis set ทำให้การคำนวณง่ายขึ้นโดยแทนออร์บิทัลอะตอมมิกเป็นชุดของฟังก์ชันที่กำหนดไว้ล่วงหน้า แทนที่จะแก้หาฟังก์ชันคลื่นต่อเนื่องโดยตรง
ทดสอบความเข้าใจ
พิจารณา trial wave function ซึ่ง คือค่าคงที่ normalization และ คือพารามิเตอร์ที่ปรับได้
(a) ทำให้ trial wave function เป็น normalize โดยหา ที่ทำให้ .
Answer
การทำ normalize สำหรับ trial wave function ที่กำหนด:
ใช้ Gaussian integral:
กำหนด จะได้:
(b) คำนวณค่าคาดหวังของ Hamiltonian ที่กำหนดโดย โดยที่ ซึ่งสอดคล้องกับศักย์ของ simple harmonic oscillator
Answer
Hamiltonian สำหรับ harmonic oscillator คือ:
ค่าคาดหวังของ kinetic energy
หาอนุพันธ์อันดับสอง:
ดังนั้น:
ใช้ผลลัพธ์ Gaussian integral มาตรฐาน:
ค่าคาดหวังของ potential energy
โดยใช้:
เราได้:
ค่าคาดหวังของพลังงานรวม
(c) ใช้หลักการ variational เพื่อหา ที่เหมาะสมที่สุดโดยการลด ให้น้อยที่สุด
Answer
ปรับค่า เพื่อให้พลังงานต่ำที่สุด
หาอนุพันธ์
แก้:
แทนค่า ลงใน :
ซึ่งตรงกับพลังงาน ground state ของ quantum harmonic oscillator อย่างแม่นยำ
VQE (Variational Quantum Eigensolver)
Variational quantum eigensolver (VQE) คือวิธีหลักที่เราจะใช้ในการสำรวจกระบวนการ และที่นี่เราจะมาดูกันว่า VQE คืออะไรและทำงานอย่างไร แต่ก่อนอื่น ลองหยุดพักและดูสิ่งสำคัญมากอย่างหนึ่งผ่านคำถามตรวจสอบความเข้าใจ
ทดสอบความเข้าใจ
ถ้าเรามีกลยุทธ์สำหรับปัญหาทางเคมีมากมายอยู่แล้ว แล้วทำไมเราจึงต้องใช้คอมพิวเตอร์ควอนตัม? และจุดประสงค์ของการใช้ทั้งคอมพิวเตอร์ควอนตัมและคลาสสิกร่วมกันคืออะไร?
Answer
การคำนวณเชิงควอนตัมมีโอกาสที่จะปฏิวัติวงการเคมีโดยจัดการกับปัญหาที่คอมพิวเตอร์คลาสสิกต่อสู้ดิ้นรน เนื่องจากการ scaling แบบ exponential ของสถานะควอนตัม Richard Feynman กล่าวไว้อย่างโด่งดังว่า เพื่อจำลองธรรมชาติ การคำนวณก็ต้องเป็นควอนตัมด้วย [อ้างอิง 1]
ตัวอย่างเช่น การจำลองคาเฟอีนด้วย basis set ที่ง่ายที่สุด (STO-3G) จะต้องใช้ bits ซึ่งใหญ่กว่าจำนวนดาวทั้งหมดในจักรวาลที่สังเกตได้ () มาก [อ้างอิง 2] คอมพิวเตอร์ควอนตัมสามารถอธิบายออร์บิทัลอิเล็กตรอนของคาเฟอีนด้วยเพียง 160 qubits
คอมพิวเตอร์ควอนตัมประมวลผลปฏิสัมพันธ์ควอนตัมโดยธรรมชาติโดยใช้ superposition และ entanglement ซึ่งเป็นแนวทางที่มีแนวโน้มดีในการทำให้การจำลองโมเลกุลมีความแม่นยำ นอกจากนี้ เรายังสามารถรวมข้อดีของทั้งคอมพิวเตอร์ควอนตัม (การจำลองอิเล็กตรอน) และคอมพิวเตอร์คลาสสิก (การประมวลผลข้อมูลก่อน/หลัง การจัดการกระบวนการอัลกอริทึม การปรับค่า และอื่นๆ) เข้าด้วยกัน สิ่งเหล่านี้คาดว่าจะช่วยเพิ่มประสิทธิภาพการค้นพบวัสดุใหม่ การออกแบบยา และการทำนายปฏิกิริยา ลดการทดลองแบบลองผิดลองถูกที่มีต้นทุนสูง [อ้างอิง 3][อ้างอิง 4]
หากต้องการทราบว่าทำไมจึงต้องใช้คอมพิวเตอร์ควอนตัมสำหรับปัญหาทางเคมี และทำไมต้องใช้ทั้งทรัพยากรการคำนวณเชิงควอนตัมและคลาสสิก ดูบทความต่อไปนี้:
ตอนนี้กลับมาที่ VQE กัน
VQE รวมพลังของคอมพิวเตอร์ควอนตัมเข้ากับคอมพิวเตอร์คลาสสิก โดยพื้นฐานแล้วใช้หลักการ variational เพื่อหา ground state energy ของระบบ เพื่อทำความเข้าใจ VQE ให้แบ่งออกเป็นสามส่วนก่อน:
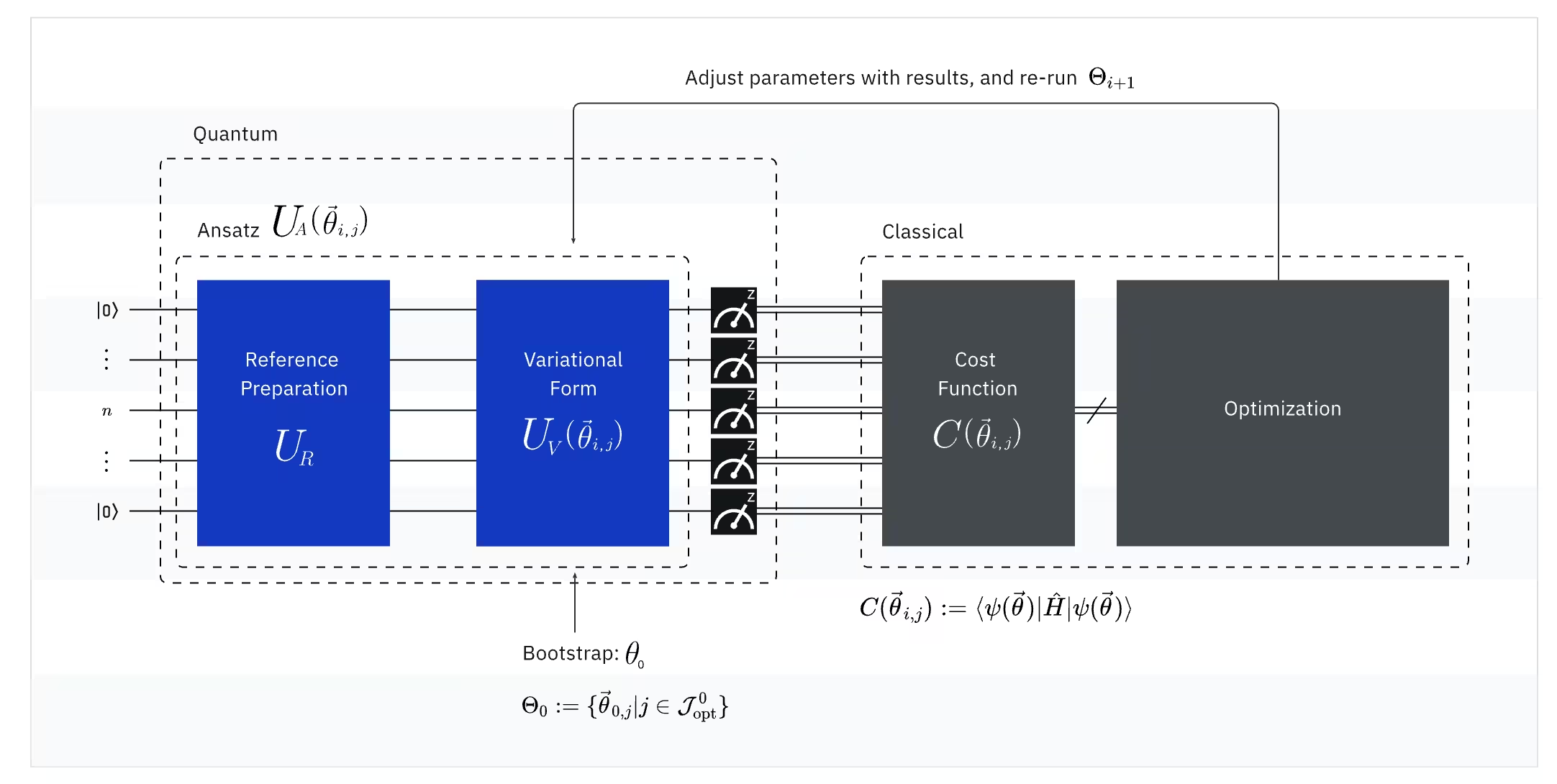
(ควอนตัม) Observable: Molecular Hamiltonian (พลังงานของโมเลกุล)
ใน VQE Hamiltonian ของโมเลกุล/อะตอมเป็น observable ซึ่งหมายความว่าเราสามารถวัดค่าของมันผ่านการทดลองได้ เป้าหมายของเราคือการหาพลังงานต่ำสุดที่เป็นไปได้ (ground state energy) ของโมเลกุล ในการทำเช่นนี้ เราใช้ trial quantum state ที่สร้างโดย parameterized quantum circuit (ansatz) เราวัด observable และปรับค่า quantum state จนกว่าจะถึงพลังงานต่ำสุดที่เป็นไปได้
Basis set ที่ใช้สำหรับ molecular Hamiltonian กำหนดจำนวน qubits ที่จำเป็น และส่งผลโดยตรงต่อความแม่นยำของ VQE การเลือก basis set ที่เหมาะสมมีความสำคัญอย่างยิ่งในการสร้างสมดุลระหว่างประสิทธิภาพและความแม่นยำ เพื่อลดความซับซ้อนในการคำนวณโดยไม่เปลี่ยน basis set เราสามารถใช้กลยุทธ์อย่างการบังคับใช้ symmetry และ active space reduction โมเลกุลหลายชนิดมีรูปร่างสมมาตร (เช่น ผีเสื้อหรือเกล็ดหิมะ) ซึ่งหมายความว่าบางส่วนมีพฤติกรรมเหมือนกัน แทนที่จะคำนวณทุกอย่างแยกกัน เราสามารถมุ่งเน้นเฉพาะส่วนที่เป็นเอกลักษณ์ ประหยัดทรัพยากรควอนตัม ซึ่งเป็นการใช้ประโยชน์จาก symmetry ใน active space reduction เราพิจารณาเฉพาะออร์บิทัลที่สำคัญ เนื่องจากไม่ใช่อิเล็กตรอนทุกตัวที่ส่งผลกระทบอย่างมีนัยสำคัญต่อพลังงานโมเลกุล อิเล็กตรอนที่อยู่ใกล้นิวเคลียสส่วนใหญ่จะไม่เปลี่ยนแปลง ในขณะที่บางตัวมีอิทธิพลต่อการสร้างพันธะ ด้วยการใช้วิธีการเหล่านี้ เราสามารถทำให้ VQE มีประสิทธิภาพมากขึ้นในขณะที่รักษาความแม่นยำไว้
เมื่อเราได้ molecular Hamiltonian โดยใช้ basis set ที่เหมาะสมและกลยุทธ์ข้างต้นแล้ว เราจำเป็นต้องแปลง Hamiltonian นี้ให้เหมาะสมสำหรับคอมพิวเตอร์ควอนตัม การ mapping ปัญหาไปยังตัวดำเนินการ Pauli อาจค่อนข้างซับซ้อน โดยเฉพาะในเคมีควอนตัม ซึ่งทำงานกับอนุภาคที่แยกแยะไม่ได้ (อิเล็กตรอน) เนื่องจาก qubits สามารถแยกแยะได้ เราจะไม่ลงรายละเอียดเกี่ยวกับการ mapping ที่นี่ แต่ขอแนะนำแหล่งข้อมูลต่อไปนี้ การอภิปรายทั่วไปเกี่ยวกับการ mapping ปัญหาไปยัง quantum operators สามารถดูได้ใน Quantum computing in practice การอภิปรายโดยละเอียดเพิ่มเติมเกี่ยวกับการ mapping ปัญหาเคมีไปยัง quantum operators สามารถดูได้ใน Quantum chemistry with VQE
สำหรับโมดูลนี้ เราจะให้ Hamiltonian แบบ one-qubit ที่เหมาะสมสำหรับ และ เพื่อให้เราสามารถมุ่งเน้นไปที่การใช้คอมพิวเตอร์ควอนตัมได้ Hamiltonian แบบ one-qubit เหล่านี้ถูกเตรียมโดยใช้ STO-6G basis set และ Jordan-Wigner mapping ซึ่งเป็นการ mapping ที่ตรงไปตรงมาที่สุดพร้อมการตีความทางกายภาพที่ง่ายที่สุด เนื่องจากมัน map การครอบครองของ spin-orbital หนึ่งไปยังการครอบครองของ qubit หนึ่ง นอกจากนี้ เราใช้ เทคนิคการลด qubit โดยใช้ symmetry ของ Hamiltonian ซึ่งใช้รูปแบบในการที่ spin occupation มีพฤติกรรมเพื่อลดจำนวน qubits สำหรับโมเลกุล เราสมมติว่าระยะห่างระหว่างอะตอมไฮโดรเจนสองอะตอมคือ 0.735
(ควอนตัม) Ansatz: Trial wave function (วิธีสร้าง quantum state เริ่มต้นด้วย quantum circuit)
สำหรับ VQE ansatz (พหูพจน์: ansätze) ประกอบด้วยองค์ประกอบหลักสองส่วน ส่วนแรกคือ initial state preparation ซึ่งตั้งค่าสถานะของ qubit โดยการใช้ quantum gates ที่ไม่มีพารามิเตอร์แบบ variational ส่วนที่สองคือ parameterized quantum circuit ซึ่งเป็น quantum circuit พิเศษที่มีพารามิเตอร์ที่ปรับได้ คล้ายกับปุ่มหมุนบนวิทยุ พารามิเตอร์เหล่านี้จะถูกใช้สำหรับส่วนสุดท้าย — classical optimizer — เพื่อช่วยให้เราถึง ground state ที่ดีที่สุด
ในส่วนหลักการ variational เราได้เรียนรู้ว่าคุณภาพของ trial state ส่งผลต่อคุณภาพของผลลัพธ์ของอัลกอริทึม variational ซึ่งหมายความว่าการเลือก ansatz ที่ดีมีความสำคัญใน VQE อีกครั้ง นี่เป็นหัวข้อที่หลากหลายและซับซ้อน เราจะไม่ครอบคลุมประเภทต่างๆ ของ ansatz หรือต้นกำเนิดของพวกมันที่นี่ หากสนใจเรียนรู้เพิ่มเติมเกี่ยวกับ parameterized quantum circuits และ ansatz คุณสามารถสำรวจบทเรียน Ansatz and variational form จากคอร์ส Variational algorithm design ซึ่งให้คำอธิบายและตัวอย่างโดยละเอียดของ ansätze
เนื่องจากเราจะใช้ Hamiltonian แบบ one-qubit ในโมดูลนี้ เราจึงต้องการ parameterized quantum circuit แบบ one-qubit เป็น ansatz เราจะเห็น ansätze แบบ one-qubit สามประเภทในส่วนต่อไป เราจะเปรียบเทียบพวกมันและอภิปรายข้อพิจารณาสำคัญในการเลือก ansatz
(คลาสสิก) Optimizer: การปรับแต่ง quantum circuit อย่างละเอียด
เมื่อคอมพิวเตอร์ควอนตัมวัดพลังงานของ observable จาก ansatz แล้ว พารามิเตอร์ของ ansatz และค่าพลังงานจะถูกส่งไปยัง classical optimizer เพื่อปรับแต่ง กระบวนการปรับค่านี้ดำเนินการบนคอมพิวเตอร์คลาสสิก โดยทั่วไปใช้แพ็กเกจวิทยาศาสตร์ทั่วไปอย่าง SciPy
Classical optimizer ถือว่าพลังงานที่วัดได้เป็น cost function ในปัญหาการปรับค่า cost function (บางครั้งเรียกว่า objective function) คือฟังก์ชันทางคณิตศาสตร์ที่วัดว่าวิธีแก้ปัญหาหนึ่งๆ "ดี" แค่ไหน เป้าหมายของ optimizer คือการหาชุดพารามิเตอร์ที่ทำให้ cost function นี้มีค่าน้อยที่สุด ในบริบทของการหา ground state energy ของโมเลกุล พลังงานเองทำหน้าที่เป็น cost function เราต้องการหาพารามิเตอร์สำหรับ quantum circuit ("วิธีแก้") ที่ให้พลังงานต่ำสุดที่เป็นไปได้ Classical optimizer ใช้ค่าพลังงานที่วัดได้ (ต้นทุน) นี้และกำหนดชุดพารามิเตอร์ที่ปรับค่าแล้วสำหรับ quantum ansatz ชุดถัดไป พารามิเตอร์ที่อัปเดตเหล่านี้จะถูกส่งกลับไปยัง quantum circuit และกระบวนการจะทำซ้ำ ในแต่ละการวนซ้ำ classical optimizer จะปรับพารามิเตอร์เพื่อพยายามลดพลังงาน (ลด cost function) จนกว่าจะตรงตามเกณฑ์การ convergence ที่กำหนดไว้ล่วงหน้า โดยหวังว่าจะพบพลังงานต่ำสุดที่เป็นไปได้ (สอดคล้องกับ ground state ของโมเลกุลสำหรับระยะพันธะและ basis set นั้นๆ)
มีกลยุทธ์การปรับค่ามากมายจากแพ็กเกจวิทยาศาสตร์อย่าง SciPy คุณสามารถค้นหาเพิ่มเติมได้ในบทเรียน Optimization loops ของคอร์ส Variational algorithm design ที่นี่เราจะใช้ COBYLA (Constrained Optimization BY Linear Approximations) ซึ่งเป็นอัลกอริทึมการปรับค่าที่เหมาะสมสำหรับ energy landscape ที่ซับซ้อน โดยเฉพาะอย่างยิ่ง COBYLA ไม่พยายามคำนวณ gradient ของฟังก์ชันที่กำลังศึกษา จึงเรียกว่าเป็น gradient-free optimizer ลองนึกภาพว่าคุณพยายามหายอดเขาสูงสุดในเทือกเขาโดยหลับตา เนื่องจากคุณมองไม่เห็นภูมิทัศน์ทั้งหมด คุณจึงก้าวเดินทีละน้อยในทิศทางต่างๆ ขณะตรวจสอบว่าขึ้นหรือลง COBYLA ทำงานในลักษณะเดียวกัน โดยเคลื่อนที่ผ่านพื้นที่ของพารามิเตอร์ ทดสอบค่าต่างๆ และค่อยๆ ปรับปรุงผลลัพธ์จนกว่าจะพบค่าที่ดีที่สุด
ตอนนี้คุณพร้อมแล้วที่จะทำการคำนวณ VQE ลองทำคำถามตรวจสอบความเข้าใจด้านล่าง ซึ่งสรุปกระบวนการโดยรวม
ทดสอบความเข้าใจ
เติมช่องว่างด้วยคำที่ถูกต้องเพื่อให้สรุปกระบวนการ VQE สมบูรณ์ แล้วคลิกเพื่อตรวจสอบคำตอบของคุณ
VQE คืออัลกอริทึมควอนตัมแบบ variational ที่รวมพลังของ (1) ________ และการคำนวณเชิงคลาสสิก ใช้เพื่อหา (2) __________ ของโมเลกุล กระบวนการเริ่มต้นด้วยการกำหนด (3) __________ ซึ่งแทนพลังงานรวมของระบบและทำหน้าที่เป็น observable ในการวัดควอนตัม ถัดมา เราเตรียม (4) __________ ซึ่งเป็น quantum circuit ที่มีพารามิเตอร์ปรับได้แทน trial wave function ของโมเลกุล พารามิเตอร์เหล่านี้ได้รับการปรับค่าโดยใช้ (5) __________ ซึ่งเป็นอัลกอริทึมคลาสสิกที่ปรับพารามิเตอร์ซ้ำๆ เพื่อลดพลังงานที่วัดได้ ในการอภิปรายข้างต้น เราใช้ (6) __________ optimizer ซึ่งปรับพารามิเตอร์ ansatz โดยไม่ต้องการการคำนวณ derivative กระบวนการดำเนินต่อจนกว่าเราจะถึง (7) __________ ซึ่งหมายความว่าเราได้พบพลังงานต่ำสุดที่เป็นไปได้ของโมเลกุล
คลังคำศัพท์:
- classical optimizer
- ground state energy
- hardware-efficient
- ansatz
- molecular Hamiltonian
- COBYLA
- quantum computing
- convergence
Answer
1 → quantum computing
2 → ground state energy
3 → molecular Hamiltonian
4 → ansatz
5 → classical optimizer
6 → COBYLA
7 → convergence
คำนวณ ground state energy ของอะตอมไฮโดรเจนด้วย VQE
ตอนนี้ลองใช้สิ่งที่เราได้เรียนรู้มาคำนวณ ground state energy ของอะตอมไฮโดรเจน ตลอดทั้งโมดูล เราจะใช้กรอบการทำงานสำหรับการคำนวณเชิงควอนตัมที่เรียกว่า "Qiskit patterns" ซึ่งแบ่งขั้นตอนการทำงานออกเป็นขั้นตอนต่อไปนี้:
- ขั้นตอนที่ 1: Map input เชิงคลาสสิกไปยังปัญหาเชิงควอนตัม
- ขั้นตอนที่ 2: ปรับปัญหาให้เหมาะสมสำหรับการรันเชิงควอนตัม
- ขั้นตอนที่ 3: รันโดยใช้ Qiskit Runtime primitives
- ขั้นตอนที่ 4: Post-processing และการวิเคราะห์เชิงคลาสสิก

เราจะทำตามขั้นตอนเหล่านี้เป็นหลัก
มาเริ่มต้นด้วยการโหลดแพ็กเกจที่จำเป็นบางส่วน รวมถึง Qiskit Runtime primitives นอกจากนี้เราจะเลือกคอมพิวเตอร์ควอนตัมที่ว่างน้อยที่สุดที่มีอยู่
โค้ดด้านล่างนี้ใช้สำหรับบันทึก credentials ในการใช้งานครั้งแรก อย่าลืมลบข้อมูลนี้ออกจาก notebook หลังจากบันทึกลงใน environment ของคุณ เพื่อป้องกันไม่ให้ credentials ถูกแชร์โดยไม่ตั้งใจเมื่อคุณแชร์ notebook ดูคำแนะนำเพิ่มเติมได้ที่ ตั้งค่าบัญชี IBM Cloud ของคุณ และ เริ่มต้นใช้งาน service ในสภาพแวดล้อมที่ไม่น่าเชื่อถือ
# Load the Qiskit Runtime service
from qiskit_ibm_runtime import QiskitRuntimeService
# Load the Runtime primitive and session
from qiskit_ibm_runtime import EstimatorV2 as Estimator
# Syntax for first saving your token. Delete these lines after saving your credentials.
# QiskitRuntimeService.save_account(channel='ibm_quantum_platform',
# instance = '<YOUR_IBM_INSTANCE_CRN>', token='<YOUR-API_KEY>', overwrite=True, set_as_default=True)
# service = QiskitRuntimeService(channel='ibm_quantum_platform')
# Load saved credentials
service = QiskitRuntimeService()
# Use the least busy backend, or uncomment the loading of a specific backend like "ibm_brisbane".
backend = service.least_busy(operational=True, simulator=False, min_num_qubits=127)
# backend = service.backend("ibm_brisbane")
print(backend.name)
ibm_brisbane
เซลล์ด้านล่างนี้จะช่วยให้คุณสลับระหว่างการใช้ simulator หรือฮาร์ดแวร์จริงได้ตลอดทั้ง notebook เราแนะนำให้รันตอนนี้เลย:
# Load the Aer simulator and generate a noise model based on the currently-selected backend.
from qiskit_aer import AerSimulator
from qiskit_aer.noise import NoiseModel
# Alternatively, load a fake backend with generic properties and define a simulator.
noise_model = NoiseModel.from_backend(backend)
# Define a simulator using Aer, and use it in Sampler.
backend_sim = AerSimulator(noise_model=noise_model)
ขั้นตอนที่ 1: Map ปัญหาไปยัง quantum circuits และ operators
เราเริ่มการคำนวณ VQE โดยการกำหนด Hamiltonian สำหรับโมเลกุลไฮโดรเจน () ที่ระยะพันธะที่กำหนด Hamiltonian นี้แทนพลังงานรวมของระบบในรูปของ qubit operators โดยได้รับการสร้างและ map จากระบบโมเลกุลโดยใช้ขั้นตอนมาตรฐาน: 1) ใช้ STO-6G basis set (ชุดเฉพาะของฟังก์ชันทางคณิตศาสตร์ที่ใช้ประมาณออร์บิทัลอิเล็กตรอน), 2) ใช้ Jordan-Wigner mapping (เทคนิคในการแปล fermionic operators ที่อธิบายอิเล็กตรอนไปยัง qubit operators) และ 3) ทำ qubit reduction โดยใช้ parity ของ Hamiltonian เพื่อลดความซับซ้อนของปัญหา
ดังที่เราอธิบายไว้ก่อนหน้านี้ ground state energies ที่คำนวณได้ขึ้นอยู่กับการเลือก basis set และ molecular geometry (เช่น ระยะพันธะ) อย่างมาก สำหรับการกำหนดค่าเฉพาะนี้และหลังจากการแปลงเหล่านี้ qubit Hamiltonian ที่ได้มีความเรียบง่าย:
ที่นี่ แทนตัวดำเนินการ identity และ แทนตัวดำเนินการ Pauli-Z ซึ่งทำงานกับ qubit เดียว สัมประสิทธิ์ได้มาจาก integrals ที่คำนวณโดยใช้ STO-6G basis set ที่ระยะพันธะนี้พร้อมการแปลงที่เหมาะสม
เมื่อกำหนด Hamiltonian นี้แล้ว เราสามารถใช้ VQE เพื่อคำนวณ ground state energy ของมัน มีประโยชน์ที่จะเปรียบเทียบ ground state energy ที่เราคำนวณได้กับค่าที่คาดหวัง สำหรับอะตอมไฮโดรเจนเดี่ยวที่แยกโดด (H) ground state energy คือ -0.5 Hartree (ในกรณีที่ไม่มีผลเชิงสัมพัทธภาพ) มาคำนวณ ground state energy ที่แม่นยำของ qubit Hamiltonian เฉพาะของเรา ตามที่กำหนดไว้ข้างต้น และเปรียบเทียบกับค่าที่รู้จักที่เกี่ยวข้อง
from qiskit.quantum_info import SparsePauliOp
import numpy as np
# Qubit Hamiltonian of the hydrogen atom generated by using STO-3G basis set and parity mapping
Hamiltonian = SparsePauliOp.from_list([("I", -0.2355), ("Z", 0.2355)])
# exact ground state energy of Hamiltonian
A = np.array(Hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print(
"The exact ground state energy of the Hamiltonian is ",
min(eigenvalues).real,
"hartree",
)
h = min(eigenvalues.real)
The exact ground state energy of the Hamiltonian is -0.471 hartree
ถัดมา เราต้องการ parameterized quantum circuit หรือ ansatz เพื่อเตรียม trial wave function สำหรับ ground state เป้าหมายคือการหาพารามิเตอร์ ที่ทำให้ค่าคาดหวังของพลังงาน มีค่าน้อยที่สุด การเลือก ansatz มีความสำคัญอย่างยิ่ง เพราะมันกำหนดชุดของ quantum states ที่ circuit ของเราสามารถเตรียมได้ ansatz ที่ "ดี" คือ ansatz ที่มีความยืดหยุ่นเพียงพอที่จะแทน state ที่ใกล้เคียงมากกับ ground state จริงของ Hamiltonian ที่เรากำลังศึกษา แต่ไม่ซับซ้อนจนต้องการพารามิเตอร์มากเกินไปหรือ circuit ที่ลึกเกินไปสำหรับคอมพิวเตอร์ควอนตัมในปัจจุบัน
ที่นี่เราจะลอง ansätze แบบ one-qubit สามแบบที่แตกต่างกัน เพื่อดูว่าอันไหนให้ "ความครอบคลุม" ที่ดีกว่าสำหรับ quantum states ที่เป็นไปได้ที่ qubit เดียวสามารถอยู่ได้ "ความครอบคลุม" หมายถึงช่วงของ quantum states ที่ ansatz circuit สามารถสร้างได้โดยการเปลี่ยนพารามิเตอร์
เราจะใช้ ansätze สามแบบตาม combination ต่างๆ ของ single-qubit rotational gates:
- Ansatz ที่มี rotational gate 1 แกน: Ansatz นี้ใช้การหมุนรอบแกนเดียวเท่านั้น () บน Bloch sphere นี่สอดคล้องกับการเคลื่อนที่ตามวงกลมเฉพาะเพียงวงเดียว นี่คือ ansatz ที่ยืดหยุ่นน้อยที่สุดและครอบคลุม states ที่จำกัด
- Ansätze ที่มี rotational gate 2 แกนสองแบบ: Ansätze เหล่านี้รวมการหมุนรอบสองแกนที่ต่างกัน ( และ ) ซึ่งช่วยให้เราเข้าถึงส่วนที่ใหญ่กว่าของ Bloch sphere เมื่อเทียบกับการหมุนรอบแกนเดียว
ด้วยการเปรียบเทียบผลลัพธ์ VQE ที่ได้จาก ansätze สามแบบนี้ เราสามารถเห็นว่าความยืดหยุ่นและความครอบคลุมของ state-space ของ ansatz ส่งผลต่อความสามารถในการหา ground state energy ที่แท้จริงของ Hamiltonian ที่ง่ายขึ้นของเราอย่างไร ansatz ที่ยืดหยุ่นมากกว่ามี ศักยภาพ ในการหาการประมาณที่ดีกว่า แต่อาจทำให้ classical optimizer ทำงานยากขึ้นด้วย
from qiskit import QuantumCircuit
from qiskit.circuit import Parameter
from qiskit.quantum_info import Statevector, DensityMatrix, Pauli
theta = Parameter("θ")
phi = Parameter("φ")
lam = Parameter("λ")
ansatz1 = QuantumCircuit(1)
ansatz1.rx(theta, 0)
ansatz2 = QuantumCircuit(1)
ansatz2.rx(theta, 0)
ansatz2.rz(phi, 0)
ansatz3 = QuantumCircuit(1)
ansatz3.rx(theta, 0)
ansatz3.rz(phi, 0)
ansatz3.rx(lam, 0)
<qiskit.circuit.instructionset.InstructionSet at 0x1059def80>
ตอนนี้มาสร้างตัวเลขสุ่ม 5000 ตัวสำหรับแต่ละพารามิเตอร์และพล็อตการกระจายของ quantum states แบบสุ่มที่สร้างโดย ansätze สามแบบด้วยพารามิเตอร์สุ่มเหล่านี้ คุณสามารถคิดว่าพารามิเตอร์เหล่านี้เหมือนการหมุนรอบแกนต่างๆ บนพื้นผิวทรงกลม เพื่อดูการกระจายของ quantum state เราจะใช้ Bloch Sphere ซึ่งเป็นทรงกลมสามมิติที่แสดงสถานะของ qubit เดียว จุดใดๆ บนทรงกลมแทน state ที่เป็นไปได้ของ qubit โดยขั้วเหนือและขั้วใต้เหมือนกับ "0" และ "1" เชิงคลาสสิก แต่ qubit ยังสามารถอยู่ที่ใดก็ได้ระหว่างนั้น แสดงคุณสมบัติควอนตัมพิเศษอย่าง superposition ก่อนอื่น เตรียมฟังก์ชันที่จำเป็นเพื่อพล็อต 3D Bloch sphere และเตรียมพารามิเตอร์สุ่ม 5000 ชุด
import matplotlib.pyplot as plt
def plot_bloch(bloch_vectors):
# Extract X, Y, Z coordinates for 3D projection
X_coords = bloch_vectors[:, 0]
Z_coords = bloch_vectors[:, 2]
# Compute Y coordinates from X and Z to approximate the full Bloch sphere projection
Y_coords = bloch_vectors[:, 1]
# Create 3D plot
fig = plt.figure(figsize=(8, 8))
ax = fig.add_subplot(111, projection="3d")
ax.scatter(X_coords, Y_coords, Z_coords, color="blue", alpha=0.6)
# Labels and title
ax.set_xlabel("X")
ax.set_ylabel("Y")
ax.set_zlabel("Z")
ax.set_title("Parameterized 1-Qubit Circuit on 3D Bloch Sphere")
# Set axis limits and make them equal
ax.set_xlim([-1, 1])
ax.set_ylim([-1, 1])
ax.set_zlim([-1, 1])
# Ensure equal aspect ratio for all axes
ax.set_box_aspect([1, 1, 1]) # Equal scaling for x, y, z axes
# Show grid
ax.grid(True)
plt.show()
num_samples = 5000 # Number of random states
theta_vals = np.random.uniform(0, 2 * np.pi, num_samples)
phi_vals = np.random.uniform(0, 2 * np.pi, num_samples)
lam_vals = np.random.uniform(0, 2 * np.pi, num_samples)
ลองดูว่า ansatz แรกของเราทำงานอย่างไร
# List to store Bloch Sphere XZ coordinates
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create a circuit and bind parameters
qc = ansatz1
bound_qc = qc.assign_parameters({theta: theta_vals[i]}) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to a numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
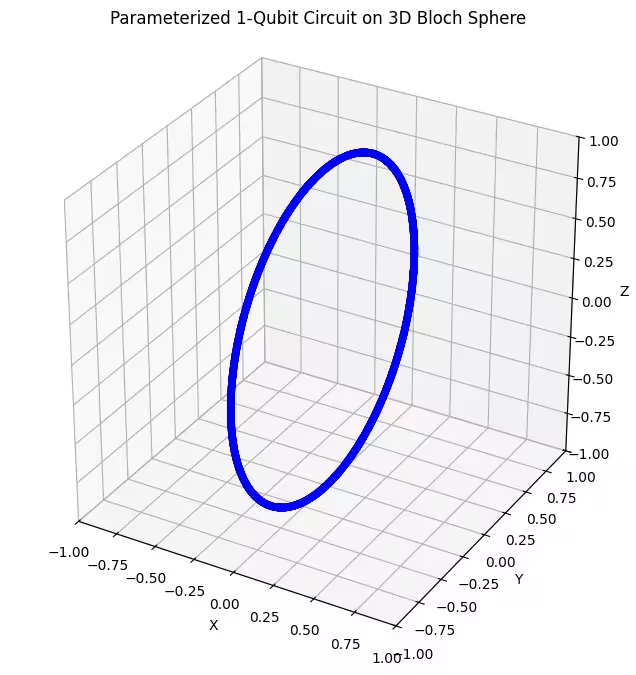
เราสามารถเห็นว่า ansatz แรกของเราคืน quantum states ที่กระจายเป็นรูปวงแหวนบน Bloch sphere ซึ่งสมเหตุสมผล เพราะเราให้พารามิเตอร์การหมุนเพียงตัวเดียวกับ ansatz มันจึงสามารถสร้าง states ที่หมุนรอบแกนเดียวเท่านั้น การเริ่มต้นจากจุด และหมุนรอบแกนเดียวจะให้วงแหวนเสมอ ต่อไปลองตรวจสอบ ansatz ที่สองของเรา ซึ่งมี rotational gates ตั้งฉากสองตัว ได้แก่ Rx และ Rz
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz2
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i]}
) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
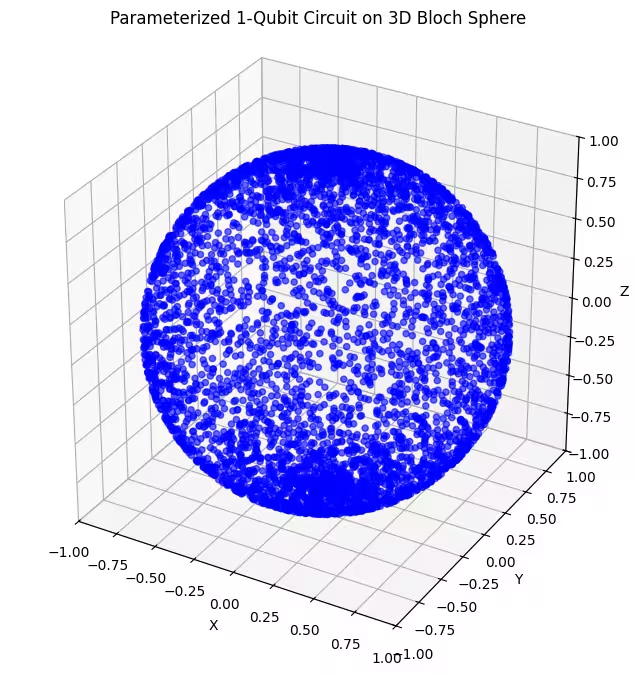
ที่นี่เราสามารถเห็นว่า ansatz ที่สองของเราครอบคลุมพื้นที่ Bloch sphere ได้มากขึ้น แต่สังเกตว่าจุดมีความหนาแน่นมากขึ้นรอบขั้วและกระจายมากขึ้นรอบเส้นศูนย์สูตร ตอนนี้ถึงเวลาตรวจสอบ ansatz สุดท้ายของเรา
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz3
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i], lam: lam_vals[i]}
)
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
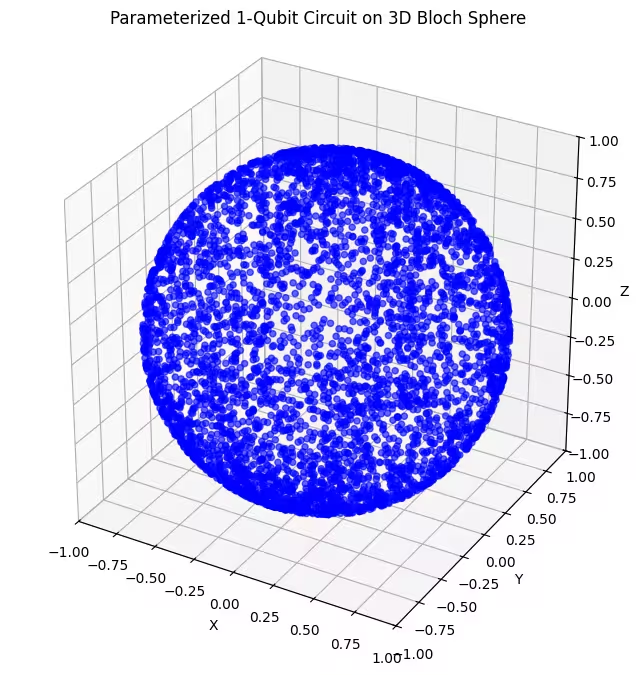
ที่นี่คุณสามารถเห็น quantum states ที่กระจายสม่ำเสมอมากขึ้นที่สร้างโดย ansatz สุดท้ายของเรา
ดังที่กล่าวไว้ สิ่งที่ดีที่สุดที่ควรทำคือการเรียนรู้เกี่ยวกับ ground state ที่คุณกำลังมองหา และใช้ ansatz ที่เหมาะสมในการสำรวจ states ที่ใกล้เคียงกับ ground state นั้น ตัวอย่างเช่น ถ้าเรารู้ว่า ground state ของเราอยู่ใกล้ขั้ว เราอาจเลือก ansatz 2 เพื่อความเรียบง่าย เราจะใช้ ansatz 3 ซึ่งสำรวจ Bloch sphere ทั้งหมดอย่างสม่ำเสมอ
ตอนนี้ที่เราเลือก ansatz แล้ว มาวาด circuit กัน
# Pre-defined ansatz circuit and operator class for Hamiltonian
ansatz = ansatz3
num_params = ansatz.num_parameters
print("This circuit has ", num_params, "parameters")
ansatz.draw("mpl", style="iqp")
This circuit has 3 parameters

ขั้นตอนที่ 2: ปรับให้เหมาะสมสำหรับฮาร์ดแวร์เป้าหมาย
เมื่อรันการคำนวณบนคอมพิวเตอร์ควอนตัมจริง เราไม่ได้สนใจแค่ logic ของ quantum circuit เราต้องสนใจด้วยว่าคอมพิวเตอร์ควอนตัมนั้นๆ สามารถดำเนินการอะไรได้บ้าง และ qubits ที่เราใช้อยู่ที่ไหนบนคอมพิวเตอร์ควอนตัม พวกมันอยู่ชิดกันไหม? หรืออยู่ห่างกัน? ดังนั้น ขั้นตอนถัดไปคือการเขียน circuit ใหม่โดยใช้ gates ที่เป็นธรรมชาติสำหรับคอมพิวเตอร์ควอนตัมที่เราจะใช้ และคำนึงถึง qubit layout ด้วย ซึ่งทำได้โดย transpilation — หลังจากกระบวนการนี้ คุณจะเห็น ansatz ง่ายๆ ของเราถูกแปลงเป็น gates ชุดอื่น และ qubits เชิงนามธรรมของเราจะถูก map ไปยัง physical qubits บนคอมพิวเตอร์ควอนตัมจริง
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
config = backend.configuration()
print("Backend: {config.backend_name}")
print("Native gates: ", config.supported_instructions, ",")
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa = pm.run(ansatz)
ansatz_isa.draw(output="mpl", idle_wires=False, style="iqp")
Backend: {config.backend_name}
Native gates: ['ecr', 'id', 'delay', 'measure', 'reset', 'rz', 'sx', 'x'] ,

คุณจะเห็นว่า gates rx, rz ของ ansatz ถูกแปลงเป็นชุดของ gates rz, sx ซึ่งเป็น native gates ของ backend ของเรา นอกจากนี้ คุณจะเห็นว่า q0 ของเราถูก map ไปยัง physical qubit ที่ห้า เราต้อง map Hamiltonian ตามการเปลี่ยนแปลงเหล่านี้ด้วย ดังในโค้ดต่อไปนี้:
Hamiltonian_isa = Hamiltonian.apply_layout(layout=ansatz_isa.layout)
ขั้นตอนที่ 3: รันบนฮาร์ดแวร์เป้าหมาย
ตอนนี้ถึงเวลารัน VQE บน QPU จริงแล้ว สำหรับสิ่งนี้ อันดับแรกเราต้องการ cost function สำหรับกระบวนการปรับค่า ซึ่งประเมินค่าคาดหวังของ Hamiltonian กับ quantum state ที่สร้างโดย ansatz ไม่ต้องกังวล คุณไม่จำเป็นต้องเขียนโค้ดทุกอย่างเอง เราเตรียมฟังก์ชันนี้ไว้ให้แล้ว และสิ่งที่คุณต้องทำคือรันเซลล์ด้านล่าง
def cost_func(params, ansatz, hamiltonian, estimator):
"""Return estimate of energy from estimator
Parameters:
params (ndarray): Array of ansatz parameters
ansatz (QuantumCircuit): Parameterized ansatz circuit
hamiltonian (SparsePauliOp): Operator representation of Hamiltonian
estimator (EstimatorV2): Estimator primitive instance
cost_history_dict: Dictionary for storing intermediate results
Returns:
float: Energy estimate
"""
pub = (ansatz, [hamiltonian], [params])
result = estimator.run(pubs=[pub]).result()
energy = result[0].data.evs[0]
cost_history_dict["iters"] += 1
cost_history_dict["prev_vector"] = params
cost_history_dict["cost_history"].append(energy)
print(f"Iters. done: {cost_history_dict['iters']} [Current cost: {energy}]")
return energy
สุดท้าย เราเตรียม initial parameters สำหรับ ansatz และกระบวนการปรับค่า คุณสามารถใช้ค่าศูนย์ทั้งหมดหรือค่าสุ่มได้ เราได้เลือก initial parameters ด้านล่างนี้ แต่รู้สึกอิสระที่จะ comment หรือ uncomment บรรทัดต่างๆ ในเซลล์เพื่อสุ่มพารามิเตอร์แบบ uniform จาก 0 ถึง
# x0 = np.random.uniform(0, 2*pi, 3)
x0 = [1, 1, 0]
# QPU Est. 2min for ibm_brisbane
from scipy.optimize import minimize
from qiskit_ibm_runtime import Batch
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, Hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 10, "tol": 0.01},
)
batch.close()
Iters. done: 1 [Current cost: -0.3361517318448143]
Iters. done: 2 [Current cost: -0.4682546422099432]
Iters. done: 3 [Current cost: -0.38985802144149584]
Iters. done: 4 [Current cost: -0.38319217316749354]
Iters. done: 5 [Current cost: -0.4628720756579032]
Iters. done: 6 [Current cost: -0.4683301936226905]
Iters. done: 7 [Current cost: -0.45480498699294747]
Iters. done: 8 [Current cost: -0.4690533242050814]
Iters. done: 9 [Current cost: -0.465867415110354]
Iters. done: 10 [Current cost: -0.4606882723137227]
h_vqe = res.fun
print("The reference ground state energy is ", min(eigenvalues))
print("The computed ground state energy is ", h_vqe)
The reference ground state energy is (-0.471+0j)
The computed ground state energy is -0.4690533242050814
ยินดีด้วย! คุณเพิ่งทำการทดลองเคมีควอนตัมครั้งแรกสำเร็จแล้ว เราสามารถเห็นความแตกต่างระหว่าง ground state energy ที่แม่นยำของ Hamiltonian กับค่าของเรา แต่เนื่องจากเราใช้เทคนิค error mitigation เริ่มต้น (ซึ่งแก้ไขข้อผิดพลาดการอ่านค่า) ความแตกต่างจึงเล็กน้อย นี่เป็นจุดเริ่มต้นที่ดีมาก!
หมายเหตุ: คุณสามารถได้ผลลัพธ์ที่ดีขึ้นโดยการตั้งค่าระดับของ error mitigation โดยใช้ resilience_level ค่าเริ่มต้นคือ 1 และถ้าคุณตั้งค่าที่สูงขึ้น มันจะใช้เวลา QPU มากขึ้นแต่อาจให้ผลลัพธ์ที่ดีขึ้น
ขั้นตอนที่ 4: Post-process
ถึงเวลาแล้วที่จะดูว่า classical optimizer ของเราทำงานอย่างไร รันเซลล์ด้านล่างและดูรูปแบบการ convergence
fig, ax = plt.subplots()
x = np.linspace(0, 10, 10)
# Define the constant function
y_constant = np.full_like(x, h)
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

เราเริ่มต้นด้วยค่าเริ่มต้นที่ค่อนข้างดี จึงได้ค่าสุดท้ายที่ดีในเพียง 10 ขั้นตอน คุณสามารถเห็นยอดขนาดใหญ่และเล็ก ซึ่งเป็นลักษณะเฉพาะของ COBYLA optimizer — มันค้นหาพื้นที่เสมือนมองไม่เห็น landscape และปรับขนาดก้าวด้วยการวัดแต่ละครั้ง
ทดสอบความเข้าใจ
การสังเกตของคุณคืออะไร? ส่วนใดของกระบวนการข้างต้นที่เปิดรับการปรับปรุงเพื่อให้ได้ผลลัพธ์ที่ใกล้เคียงกับค่าทางทฤษฎีมากขึ้น หรือใกล้เคียงกับ ground state energy ที่แม่นยำของ Hamiltonian มากขึ้น? มีสิ่งใดบ้างที่ควรพิจารณา?
Answer
สิ่งแรกที่ควรพิจารณาคือการเปลี่ยนชุด basis ที่ใช้ในการคำนวณ Hamiltonian ของโมเลกุล ดังที่กล่าวไว้ก่อนหน้านี้ ground state energy ของอะตอม H คือ -0.5 Hartree ตามที่รู้กันดี และ STO-6G basis ที่เราเลือกไม่เพียงพอที่จะหาค่านี้ได้อย่างแม่นยำ
การเลือก basis ประเภทที่ซับซ้อนมากขึ้นจะเพิ่มจำนวน qubits ที่ใช้โดย Hamiltonian ดังนั้นเราจึงต้องเลือก ansatz ที่ซับซ้อนและเหมาะสมกว่าสำหรับปัญหาทางเคมี
สิ่งต่อไปที่ควรปรับให้เหมาะสมคือการจัดการ noise ใน QPU เทคนิค error mitigation ที่ก้าวหน้าขึ้นให้ผลลัพธ์ที่ดีขึ้นแต่อาจใช้เวลานานขึ้น นอกจากนี้ควรพิจารณาว่า shot_number ส่งผลต่อผลลัพธ์อย่างไร
สุดท้าย ประสิทธิภาพการ convergence ที่ดีขึ้นยังสามารถทำได้โดยการลอง optimizers ต่างๆ
คำนวณ ground state energy ของโมเลกุลไฮโดรเจนด้วย VQE
ตอนนี้ที่เราได้ดูกระบวนการโดยรวมของ VQE โดยใช้อะตอม แล้ว เราจะมาคำนวณ ground state energy ของโมเลกุล ให้เร็วขึ้น
ขั้นตอนที่ 1: Map ปัญหาไปยัง quantum circuits และ operators
ที่นี่เราก็ให้ Hamiltonian แบบ one-qubit ที่ใช้ STO-6G basis และ Jordan-Wigner transformation พร้อมการลด qubit โดยใช้ symmetry ของ Hamiltonian ด้วย โปรดสังเกตว่าเราใช้ระยะห่างระหว่างอะตอมไฮโดรเจนสองอะตอมที่ 0.735
ต่างจากการคำนวณอะตอมไฮโดรเจนเดี่ยว () เพื่อคำนวณ ground state ของโมเลกุลไฮโดรเจน () เราต้องคำนึงถึงแรงผลักที่กระทำระหว่างนิวเคลียสของอะตอมไฮโดรเจนสองอะตอมด้วย นอกเหนือจากพลังงานที่เกี่ยวข้องกับออร์บิทัลอิเล็กตรอน ในขั้นตอนนี้ เราจะให้ค่านี้เป็นค่าคงที่ และเราจะคำนวณค่านี้จริงๆ ในคำถามตรวจสอบความเข้าใจ
h2_hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
# exact ground state energy of hamiltonian
nuclear_repulsion = 0.71997
A = np.array(h2_hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print("Electronic ground state energy (Hartree): ", min(eigenvalues).real)
print("Nuclear repulsion energy (Hartree): ", nuclear_repulsion)
print(
"Total ground state energy (Hartree): ", min(eigenvalues).real + nuclear_repulsion
)
h2 = min(eigenvalues).real + nuclear_repulsion
Electronic ground state energy (Hartree): -1.8659468547627318
Nuclear repulsion energy (Hartree): 0.71997
Total ground state energy (Hartree): -1.1459768547627318
ขั้นตอนที่ 2: ปรับให้เหมาะสมสำหรับฮาร์ดแวร์เป้าหมาย
เนื่องจากจำนวน qubits ที่ใช้โดย VQE ก่อนหน้าและ Hamiltonian เหมือนกับ backend ที่จะใช้สำหรับการรัน เราจะใช้ ansatz ที่มีอยู่และรูปแบบที่ปรับค่าแล้วของมัน
h2_hamiltonian_isa = h2_hamiltonian.apply_layout(layout=ansatz_isa.layout)
ขั้นตอนที่ 3: รันบนฮาร์ดแวร์เป้าหมาย
ตอนนี้ถึงเวลาทำการคำนวณบน QPU จริง เกือบทุกอย่างเหมือนกัน แต่เราจะใช้ initial point ที่เหมาะสมสำหรับ Hamiltonian นอกจากนี้ ในส่วนที่วนซ้ำ การตั้งค่าบางอย่างของ Estimator ซึ่งใช้คำนวณค่าคาดหวังของ Hamiltonian สำหรับ ansatz ใน QPU จะถูกตั้งค่าแตกต่างจากการคำนวณก่อนหน้าเล็กน้อย เราจะอภิปรายการเปลี่ยนแปลงนี้เพิ่มเติมในคำถามตรวจสอบความเข้าใจ
x0 = [2, 0, 0]
# QPU time 4min for ibm_brisbane
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, h2_hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 15},
)
batch.close()
Iters. done: 1 [Current cost: -0.710621837568328]
Iters. done: 2 [Current cost: -0.2603208441168329]
Iters. done: 3 [Current cost: -0.25548711201326424]
Iters. done: 4 [Current cost: -0.581129450619904]
Iters. done: 5 [Current cost: -1.722920997605439]
Iters. done: 6 [Current cost: -1.6633324849371915]
Iters. done: 7 [Current cost: -1.8066989598929164]
Iters. done: 8 [Current cost: -1.8051093803839542]
Iters. done: 9 [Current cost: -1.802692217571555]
Iters. done: 10 [Current cost: -1.8233585485263144]
Iters. done: 11 [Current cost: -1.6904116652617205]
Iters. done: 12 [Current cost: -1.8245120321245392]
Iters. done: 13 [Current cost: -1.6837021361383608]
Iters. done: 14 [Current cost: -1.8166632606115467]
Iters. done: 15 [Current cost: -1.863446212658907]
h2_vqe = res.fun + nuclear_repulsion
print(
"The reference ground state energy is ", min(eigenvalues).real + nuclear_repulsion
)
print("The computed ground state energy is ", h2_vqe)
The reference ground state energy is -1.1459768547627318
The computed ground state energy is -1.143476212658907
แม้ว่า VQE ในทางทฤษฎีจะให้ขอบเขตบนสำหรับ ground state energy จริง แต่การใช้งานจริงบนฮาร์ดแวร์ควอนตัมจริงหรือแบบมี noise รวมถึงการประมาณที่ทำในการเตรียม Hamiltonian (เช่น basis sets หรือ qubit reduction) อาจทำให้เกิดข้อผิดพลาดที่บางครั้งส่งผลให้พลังงานที่วัดได้ต่ำกว่าค่าทางทฤษฎีที่แม่นยำหรือค่าอ้างอิงตัวเลขเฉพาะเล็กน้อย แม้จะมีข้อผิดพลาดบ้าง ผลลัพธ์ดูน่าพอใจ โดยเฉพาะอย่างยิ่งเมื่อพิจารณาจากจำนวนขั้นตอนที่น้อย ตอนนี้มาสรุปการคำนวณ VQE นี้โดยดูว่า optimizer ทำงานอย่างไร
ขั้นตอนที่ 4: Post-process
fig, ax = plt.subplots()
x = np.linspace(0, 5, 15)
# Define the constant function
y_constant = np.full_like(x, min(eigenvalues))
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

ทดสอบความเข้าใจ
มาคำนวณพลังงานผลักของนิวเคลียสของโมเลกุล ซึ่งเราใส่ไว้เป็นค่าคงที่ (0.71997 Hartree)

ใช้ กฎของ Coulomb และ atomic unit เพื่อให้แน่ใจว่าคุณได้ค่าเป็น Hartree
Answer
เนื่องจากนิวเคลียสไฮโดรเจนทั้งสองมีประจุบวก พวกมันจึงผลักกันเนื่องจากแรงไฟฟ้าสถิต การผลักนี้อธิบายด้วยกฎของ Coulomb:
โดยที่ คือประจุของ proton, คือ vacuum permittivity และ คือระยะห่างระหว่างนิวเคลียสสองอะตอม วัดเป็นเมตรหรือ Bohr radii ในหน่วยของจูล (J)
ในการคำนวณพลังงานนี้เป็น Hartrees เราต้องแปลงสมการข้างต้นเป็นระบบ Atomic Unit (AU) ใน AU, , และ Bohr radius () คือ 1 และกลายเป็นมาตราส่วนความยาวพื้นฐานใน AU ด้วยการทำให้เรียบง่ายเหล่านี้ กฎของ Coulomb ลดลงเป็น:
โดยที่ ต้องวัดเป็น Bohr radii ()
ในการแปลงระยะห่างของนิวเคลียสที่กำหนดจาก เป็น เราต้องการความสัมพันธ์การแปลงนี้:
ดังนั้น จึงกลายเป็น
ดังนั้น พลังงานผลักของนิวเคลียสของ ที่กำหนดคือ
คำนวณพลังงานของปฏิกิริยา
ตอนนี้มาใช้สิ่งที่เราได้มา! คุณได้ใช้ VQE ซึ่งเป็น variational quantum eigensolver เพื่อคำนวณ ground state energy ของอะตอม และของโมเลกุล สิ่งที่เหลือคือการใช้ค่าที่คำนวณได้เพื่อหาพลังงานของปฏิกิริยาในกระบวนการ
พลังงานของปฏิกิริยาคือการเปลี่ยนแปลงพลังงานที่เกิดขึ้นเมื่อสารทำปฏิกิริยาเพื่อสร้างสารใหม่ ลองนึกภาพว่าคุณกำลังสร้างบางอย่าง บางครั้งคุณต้องใส่พลังงานเข้าไป (เช่น การต่อตัวต่อ) และบางครั้งพลังงานก็ถูกปล่อยออกมา (เช่น ลูกบอลกลิ้งลงเนิน) ในทางเคมี ปฏิกิริยาจะดูดซับพลังงาน (endothermic) หรือปล่อยพลังงาน (exothermic)
พลังงานของปฏิกิริยาในกระบวนการ สามารถคำนวณได้โดยสูตรต่อไปนี้:
ด้วยการรันเซลล์ด้านล่าง มาดูสิ่งนี้ในรูปภาพ ที่นี่เราจะใช้ค่า ground state ที่แม่นยำของ Hamiltonian แต่ละตัว และเราจะเปรียบเทียบพลังงานของปฏิกิริยาของวิธีแก้ที่แม่นยำและผลลัพธ์ VQE
# Theoretical values
E_H_theo = h.real
E_H2_theo = h2
# Experimental values
E_H_exp = h_vqe
E_H2_exp = h2_vqe
# Calculate reaction energies
E_reaction_theo = E_H2_theo - (2 * E_H_theo)
E_reaction_exp = E_H2_exp - (2 * E_H_exp)
# Set up the plot
fig, ax = plt.subplots(figsize=(8, 6))
ax.set_xlim(0, 3)
ax.set_ylim(-1.16, -0.93) # Adjust y-axis range to highlight differences
ax.set_xticks([])
ax.set_ylabel("Energy (Hartree)")
ax.set_title("H + H → H₂ Reaction Energy Diagram")
# Plot theoretical energy levels
ax.hlines(
y=2 * E_H_theo, xmin=0.5, xmax=1.3, linewidth=2, color="r", label="2H (Exact)"
)
ax.hlines(y=E_H2_theo, xmin=1.3, xmax=2, linewidth=2, color="b", label="H₂ (Exact)")
# Plot experimental energy levels
ax.hlines(
y=2 * E_H_exp,
xmin=0.5,
xmax=1.5,
linewidth=2,
color="r",
linestyle="dashed",
label="2H (VQE)",
)
ax.hlines(
y=E_H2_exp,
xmin=1.5,
xmax=2.5,
linewidth=2,
color="b",
linestyle="dashed",
label="H₂ (VQE)",
)
# Add labels
ax.text(
1,
2 * E_H_theo,
f"2H: {2*E_H_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
2,
E_H2_theo,
f"H₂: {E_H2_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
1,
2 * E_H_exp,
f"2H_VQE: {2*E_H_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
ax.text(
2,
E_H2_exp,
f"H₂_VQE: {E_H2_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
# Add arrows for reaction energy with ΔE label in the middle
mid_y_theo = (2 * E_H_theo + E_H2_theo) / 2
mid_y_exp = (2 * E_H_exp + E_H2_exp) / 2
ax.annotate(
"",
xy=(1.3, E_H2_theo),
xytext=(1.3, 2 * E_H_theo),
arrowprops=dict(arrowstyle="<->", color="g"),
)
ax.text(
1.35, mid_y_theo, f"ΔE: {E_reaction_theo:.4f}", color="g", verticalalignment="top"
)
ax.annotate(
"",
xy=(1.5, E_H2_exp),
xytext=(1.5, 2 * E_H_exp),
arrowprops=dict(arrowstyle="<->", color="g", linestyle="dashed"),
)
ax.text(
1.55,
mid_y_exp,
f"ΔE_VQE: {E_reaction_exp:.4f}",
color="g",
verticalalignment="center",
)
# Add legend
ax.legend()
plt.show()

ดังที่แสดงในรูป แม้จะมีข้อผิดพลาดบ้าง ground state energy ที่แม่นยำของ Hamiltonians และพลังงานของปฏิกิริยาที่คำนวณโดยใช้ผลลัพธ์ VQE มีความคล้ายคลึงกัน ใกล้เคียง -0.2 Hartree
ควรสังเกตที่นี่ว่าพลังงานของปฏิกิริยานี้มีค่าลบ ซึ่งหมายความว่าพลังงานถูกปล่อยออกมาผ่านกระบวนการ และโมเลกุลที่เกิดขึ้นมีพลังงานต่ำกว่าอะตอมเดี่ยวสองอะตอม 6. บทสรุป
มาสรุปสิ่งที่เราได้เรียนรู้จนถึงตอนนี้กัน
อันดับแรก เราได้ดูเทคนิคการประมาณที่สำคัญสองอย่างที่จำเป็นในการแก้ปัญหาเคมีควอนตัม ได้แก่ หลักการ variational และการเลือก basis set ซึ่งทั้งสองอย่างเป็นพื้นฐานของ VQE เราได้สำรวจหลักการ variational ด้วยตนเอง โดยคำนวณ ground state energy ของ simple harmonic oscillator
ถัดมา เราได้สำรวจ VQE ซึ่งเป็นอัลกอริทึมที่ใช้กันอย่างแพร่หลายสำหรับการคำนวณ ground state energy ของระบบควอนตัม เราได้รันโค้ดเพื่อคำนวณ ground state energies สำหรับอะตอมไฮโดรเจน () และโมเลกุลไฮโดรเจน () โดยเฉพาะอย่างยิ่ง เราได้เรียนรู้ว่าจำเป็นต้องได้ molecular Hamiltonian ที่เหมาะสมสำหรับระบบและแปลงเป็นรูปแบบที่รันได้บนคอมพิวเตอร์ควอนตัม เราได้เห็นด้วยว่า ansatz ซึ่งเป็น parameterized quantum circuit จำเป็นต้องใช้เพื่อเตรียม trial quantum states ใน VQE และเราได้อภิปรายความสำคัญของการเลือกโครงสร้าง ansatz circuit ที่เหมาะสม นอกจากนี้เราได้เรียนรู้ว่า VQE อาศัยกระบวนการปรับค่าแบบวนซ้ำโดยใช้คอมพิวเตอร์คลาสสิก เพื่อนำ quantum circuit ไปสู่การหาสถานะพลังงานต่ำสุด และได้เห็นว่ากระบวนการ convergence ทำงานอย่างไร
สุดท้าย เราได้ใช้ ground state energies ที่คำนวณได้ของ และ ที่ได้จาก VQE เพื่อคำนวณพลังงานของปฏิกิริยาสำหรับกระบวนการ
VQE เป็นอัลกอริทึมควอนตัมระยะใกล้ที่มีประสิทธิภาพ แต่สิ่งสำคัญคือต้องตระหนักถึงข้อจำกัดของมัน ประสิทธิภาพของ VQE ขึ้นอยู่กับการเลือก ansatz เป็นอย่างมาก การหา ansatz ที่สามารถเตรียมได้อย่างมีประสิทธิภาพและสามารถแทน ground state จริงได้อย่างแม่นยำจะยิ่งท้าทายมากขึ้นสำหรับโมเลกุลที่ใหญ่และซับซ้อนกว่า นอกจากนี้ ฮาร์ดแวร์ควอนตัมในปัจจุบันยังมีความไวต่อ noise ซึ่งอาจส่งผลต่อความแม่นยำของผลลัพธ์ VQE โดยเฉพาะอย่างยิ่งสำหรับ circuits ที่ลึกกว่าหรือจำนวน qubits ที่มากกว่า แม้จะมีความท้าทายเหล่านี้ VQE ก็ยังทำหน้าที่เป็นอัลกอริทึมพื้นฐาน และการวิจัยที่กำลังดำเนินอยู่กำลังสำรวจวิธีการแบบ variational ที่ซับซ้อนยิ่งขึ้นและเทคนิค error mitigation เพื่อผลักดันขอบเขตของสิ่งที่เป็นไปได้ในเคมีควอนตัมบนคอมพิวเตอร์ควอนตัมระยะใกล้ ตัวอย่างเช่น อัลกอริทึมอย่าง Sample-based Quantum Diagonalization (SQD) กำลังได้รับการพัฒนา ซึ่งใช้ประโยชน์จาก samples ที่ได้จาก quantum circuits ร่วมกับ classical diagonalization ใน subspace เพื่อปรับปรุงการประมาณพลังงานและแก้ไขข้อจำกัดบางอย่างที่ VQE เผชิญ โดยเฉพาะอย่างยิ่งในด้านประสิทธิภาพการวัดและความทนทานต่อ noise
ทบทวนและคำถาม
แนวคิดสำคัญ:
- Variational quantum algorithm คือกระบวนทัศน์การคำนวณที่คอมพิวเตอร์คลาสสิกและคอมพิวเตอร์ควอนตัมทำงานร่วมกันเพื่อแก้ปัญหา
- ใน VQE เราเริ่มต้นด้วย Hamiltonian ของระบบและ map มันไปยัง qubits เพื่อรันบนคอมพิวเตอร์ควอนตัม เราเลือก parameterized quantum circuit หรือ ansatz และทำการวัดซ้ำๆ โดยเปลี่ยนพารามิเตอร์ของ ansatz จนกว่าจะถึงค่าพลังงานต่ำสุด การค้นหาผ่านพื้นที่พารามิเตอร์ทำได้โดยใช้ classical optimizer เพื่อให้ได้ผลลัพธ์ที่ดี จำเป็นต้องเลือก ansatz ที่ดีและ optimizer ที่เหมาะสม
- พลังงานของปฏิกิริยาคือการเปลี่ยนแปลงพลังงานรวมในปฏิกิริยาเคมี กำหนดโดยความแตกต่างระหว่างพลังงานของสารตั้งต้นและผลิตภัณฑ์
จริง/เท็จ
- หลักการ variational ระบุว่าค่าคาดหวังของพลังงานสำหรับ trial wave function ใดๆ จะมากกว่าหรือเท่ากับ ground state energy จริงเสมอ
- Basis set คือชุดของฟังก์ชันที่ใช้ประมาณ quantum wave functions
- VQE คืออัลกอริทึมควอนตัมที่ใช้แก้สมการ Schrödinger อย่างแม่นยำสำหรับ Hamiltonian ที่กำหนด
- ใน VQE parameterized quantum circuit (ansatz) ถูกใช้เพื่อเตรียม trial wave functions
- การเลือก optimizer ใน VQE (ตัวอย่างเช่น COBYLA, SPSA หรือ ADAM) ไม่ส่งผลต่อคุณภาพของผลลัพธ์
Estimatorของ Qiskit ถูกใช้เพื่อคำนวณค่าคาดหวังของ Hamiltonians ใน VQE โดยตรง
คำถามปรนัย:
- จุดประสงค์ของ Hamiltonian ใน VQE คืออะไร?
- A) เพื่อสร้าง quantum states แบบสุ่ม
- B) เพื่อกำหนดพลังงานของ quantum states
- C) เพื่อปรับค่า quantum circuits
- D) เพื่อสร้าง entanglement
- เป้าหมายหลักของอัลกอริทึม VQE คืออะไร?
- A) เพื่อหา ground state energy ของ Hamiltonian
- B) เพื่อสร้าง entanglement ระหว่าง qubits
- C) เพื่อทำ Grover's search
- D) เพื่อทำลายการเข้ารหัส RSA
- มีการสร้าง quantum states กี่ states ใน notebook นี้เพื่อเปรียบเทียบ ansatz?
- A) 100
- B) 1000
- C) 5000
- D) 10,000
- ทำไมจึงต้องใช้ classical optimizer ใน VQE?
- A) เพื่อทำการวัดเชิงควอนตัม
- B) อัปเดตพารามิเตอร์ ansatz เพื่อลดพลังงาน
- C) เพื่อ entangle qubits
- D) เพื่อสร้างความสุ่มเชิงควอนตัม
- ทำไม ansatz จึงถูกออกแบบให้มีพารามิเตอร์?
- A) เพื่อเอื้อให้เตรียม quantum state
- B) เพื่อให้สามารถค้นหา quantum states ได้ในพื้นที่กว้าง
- C) เพื่อลดความซับซ้อนของ circuit
- D) เพื่อวัด eigenvalues โดยตรง
- ข้อความใดต่อไปนี้ถูกต้องที่สุดเกี่ยวกับการเลือก ansatz ที่ดี?
- A) Ansatz ต้องสร้าง states ที่กระจายสม่ำเสมอบน Bloch sphere มิฉะนั้นจะล้มเหลว
- B) Ansatz ควรถูกปรับให้เหมาะกับระบบของคุณเพื่อให้แน่ใจว่าสามารถสร้าง states ที่ใกล้เคียงกับ ground state
- C) Ansatz ควรสร้าง states แบบสุ่มโดยใช้พารามิเตอร์แบบ variational ของมัน
- D) Ansatz ที่ดีกว่าจะมีพารามิเตอร์แบบ variational มากกว่าเสมอ
(ทางเลือก) ภาคผนวก: Optimizer overhead ตามความซับซ้อนของ ansatz
VQE เผชิญกับความท้าทายที่รู้จักกันดีหลายอย่าง [อ้างอิง 6] และต่อไปนี้เกี่ยวข้องกับสิ่งที่เราได้เรียนรู้ข้างต้น
- ความท้าทายในการเลือก ansatz
มีความท้าทายโดยธรรมชาติในการเลือก variational ansatz ที่เหมาะสม Ansätze ที่ได้แรงบันดาลใจจากเคมี (เช่น UCCSD) ให้ความแม่นยำทางกายภาพแต่ต้องการ circuits ที่ลึก ในขณะที่ hardware-efficient ansätze มี circuits ที่ตื้นกว่าแต่อาจขาดการตีความทางกายภาพ นอกจากนี้ ansätze หลายอย่างยังนำ variational parameters มากเกินไปที่มีส่วนช่วยน้อยมากในการปรับปรุงความแม่นยำแต่เพิ่มความยากในการปรับค่าอย่างมีนัยสำคัญ
- ความยากในการปรับค่า
Optimization landscape ของ VQE อาจมีบริเวณที่ gradients หายไปแบบ exponential (barren plateaus) ทำให้ classical optimizers ปรับ variational parameters ได้ยาก สำหรับสิ่งนี้ นักวิจัยได้พยายามใช้ optimizers ประเภทต่างๆ ทั้งแบบที่ใช้ gradient และไม่ใช้ gradient แต่ทั้งสองแบบเผชิญกับความท้าทาย Gradient-based optimizers ทนทุกข์กับ barren plateaus ในขณะที่ gradient-free methods ต้องการการประเมินฟังก์ชันจำนวนมาก
- Optimizer overhead
ความท้าทายที่รู้จักกันดีอีกอย่างคือ optimizer overhead ซึ่งเกี่ยวข้องกับขนาดของปัญหา Quantum circuits ที่จำเป็นสำหรับ VQE เพิ่มความลึกและความซับซ้อนเมื่อขนาดปัญหาเพิ่มขึ้น ซึ่งโดยทั่วไปจะเพิ่มจำนวนพารามิเตอร์ที่ต้องปรับค่าด้วย กระบวนการปรับค่าจะจัดการได้ยากเมื่อจำนวนพารามิเตอร์เพิ่มขึ้น นำไปสู่การ convergence ที่ช้าลงและความยากในการหาวิธีแก้ที่เหมาะสมที่สุด
ที่นี่เราจะดูความท้าทายเหล่านี้โดยการใช้ VQE สำหรับโมเลกุล กับ ansätze สองประเภทที่แตกต่างกัน
(หมายเหตุ: สิ่งนี้อาจใช้เวลา QPU มากขึ้น ดังนั้นรู้สึกอิสระที่จะใช้ simulator หากคุณมีเวลาไม่พอ)
from qiskit.circuit import ParameterVector
num_iter = 4
alpha = ParameterVector("alpha", 3)
beta = ParameterVector("beta", 3 * num_iter)
# step1: Map problem to quantum circuits and operators
hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
ansatz_1 = ansatz3
ansatz_2 = QuantumCircuit(1)
for i in range(num_iter):
ansatz_2.rx(beta[i * 3 + 0], 0)
ansatz_2.rz(beta[i * 3 + 1], 0)
ansatz_2.rx(beta[i * 3 + 2], 0)
ansatz_1.draw("mpl")
ansatz_2.draw("mpl")

# Step 2: Optimize for target hardware
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa_1 = pm.run(ansatz_1)
ansatz_isa_2 = pm.run(ansatz_2)
hamiltonian_isa_1 = hamiltonian.apply_layout(layout=ansatz_isa_1.layout)
hamiltonian_isa_2 = hamiltonian.apply_layout(layout=ansatz_isa_2.layout)
ตอนนี้มารัน VQE ด้วย initial point ที่เป็นค่าหนึ่งทั้งหมด พร้อมจำนวนขั้นตอนสูงสุด 20 ขั้นตอน และเปรียบเทียบการ convergence ของทั้งสองการรัน
# QPU time 3m 40s for ibm_brisbane
# Step 3: Execute on target hardware
from scipy.optimize import minimize
x0 = np.ones(ansatz_1.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_1, hamiltonian_isa_1, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.8782202668652658]
Iters. done: 2 [Current cost: -0.43473160695469165]
Iters. done: 3 [Current cost: -0.4076372093159749]
Iters. done: 4 [Current cost: -1.3587839859772106]
Iters. done: 5 [Current cost: -1.774529906754082]
Iters. done: 6 [Current cost: -1.541934983115727]
Iters. done: 7 [Current cost: -1.2732403113465345]
Iters. done: 8 [Current cost: -1.820842221085785]
Iters. done: 9 [Current cost: -1.8065762857059005]
Iters. done: 10 [Current cost: -1.8126394095981146]
Iters. done: 11 [Current cost: -1.8205831886180421]
Iters. done: 12 [Current cost: -1.8086715778994924]
Iters. done: 13 [Current cost: -1.8307676638629322]
Iters. done: 14 [Current cost: -1.8177328827556327]
Iters. done: 15 [Current cost: -1.8179426218088064]
Iters. done: 16 [Current cost: -1.8109239667991088]
Iters. done: 17 [Current cost: -1.824271872489647]
Iters. done: 18 [Current cost: -1.813167587671394]
Iters. done: 19 [Current cost: -1.824647343397313]
Iters. done: 20 [Current cost: -1.8219785311686143]
# Save Cost_history as a new list
ansatz_1_history = cost_history_dict["cost_history"]
# QPU time 3m 40s for ibm_brisbane
x0 = np.ones(ansatz_2.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_2, hamiltonian_isa_2, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.738191173881188]
Iters. done: 2 [Current cost: -0.42636037194506304]
Iters. done: 3 [Current cost: -1.3503788613797374]
Iters. done: 4 [Current cost: -0.9109204349776897]
Iters. done: 5 [Current cost: -0.9060873157510835]
Iters. done: 6 [Current cost: -0.7735065414083984]
Iters. done: 7 [Current cost: -1.586889197437709]
Iters. done: 8 [Current cost: -1.659215191584943]
Iters. done: 9 [Current cost: -1.245445981794618]
Iters. done: 10 [Current cost: -1.1608385766138023]
Iters. done: 11 [Current cost: -1.1551733876027737]
Iters. done: 12 [Current cost: -1.8143337768286332]
Iters. done: 13 [Current cost: -1.2510951563756598]
Iters. done: 14 [Current cost: -1.6918311531865413]
Iters. done: 15 [Current cost: -1.8163783305531838]
Iters. done: 16 [Current cost: -1.8434877732947152]
Iters. done: 17 [Current cost: -1.8461898233304472]
Iters. done: 18 [Current cost: -1.0346471214915485]
Iters. done: 19 [Current cost: -1.8322518854150687]
Iters. done: 20 [Current cost: -1.717144678705999]
ansatz_2_history = cost_history_dict["cost_history"]
fig, ax = plt.subplots()
# Define the constant function)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_1_history,
label="Ansatz with 3 parameters",
)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_2_history,
label="Ansatz with 12 parameters",
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
plt.legend()
plt.draw()

กราฟข้างต้นแสดงให้เห็นอย่างชัดเจนว่ากระบวนการปรับค่าของ ansatz ที่มีตัวแปรมากกว่าใช้เวลานานกว่าในการถึง convergence ที่เสถียร
แทนที่จะพึ่งพา single-qubit circuits ที่เรียบง่ายและ ansatz ที่ตรงไปตรงมา ความซับซ้อนของการปรับค่าจะเพิ่มขึ้นเมื่อต้องการ quantum circuits ที่ใหญ่กว่าและ ansätze ที่มีโครงสร้างซับซ้อนกว่า ซึ่งเน้นย้ำถึงความท้าทายที่รู้จักกันดีใน VQEs: overhead ของ optimizer
นักวิจัยยังคงพัฒนาวิธีการขั้นสูงต่างๆ ที่สามารถใช้คอมพิวเตอร์ควอนตัมสำหรับปัญหาทางเคมี คุณสามารถเข้าถึงสื่อการเรียนรู้ที่หลากหลายได้ที่ IBM Quantum Learning
อ้างอิง
- [อ้างอิง 1 ] Richard P. Feynman, Simulating Physics with Computers, International Journal of Theoretical Physics, 1982.
- [อ้างอิง 2] Marov, M.Y. (2015). The Structure of the Universe. In: The Fundamentals of Modern Astrophysics. Springer, New York, NY.
- [อ้างอิง 3] How to solve difficult chemical engineering problems with quantum computing, IBM Research Blog, 2023.
- [อ้างอิง 4] Y. Cao, J. Romero and A. Aspuru-Guzik, "Potential of quantum computing for drug discovery," in IBM Journal of Research and Development, vol. 62, no. 6, pp. 6:1-6:20, 1 Nov.-Dec. 2018
- [อ้างอิง 5] Present State of Molecular Structure Calculation, REv. Mod. Phys. 32, 170, 1960
- [อ้างอิง 6] Fedorov, D.A., Peng, B., Govind, N. et al. VQE method: a short survey and recent developments. Mater Theory 6, 2 (2022)