HI-VQE Chemistry - A Qiskit Function by Qunova Computing
# Added by doQumentation — required packages for this notebook
!pip install -q qiskit-ibm-catalog qiskit-ibm-runtime
# This cell is hidden from users
from qiskit_ibm_runtime import QiskitRuntimeService
service = QiskitRuntimeService()
instance = service.active_account()["instance"]
backend_name = service.least_busy(operational=True, min_num_qubits=16).name
See the API reference
Qiskit Functions เป็นฟีเจอร์ทดลองที่มีเฉพาะสำหรับผู้ใช้ IBM Quantum® Premium Plan, Flex Plan และ On-Prem (ผ่าน IBM Quantum Platform API) Plan เท่านั้น ยังอยู่ในสถานะ preview release และอาจมีการเปลี่ยนแปลง
Package versions
The code on this page was developed using the following requirements. We recommend using these versions or newer.
qiskit-ibm-runtime~=0.45.0
ภาพรวม
ในเคมีควอนตัม ปัญหาโครงสร้างอิเล็กตรอนเน้นที่การหาคำตอบของสมการ Schrödinger อิเล็กทรอนิกส์ ซึ่งเป็นฟังก์ชันคลื่นควอนตัมที่อธิบายพฤติกรรมของอิเล็กตรอนในระบบ ฟังก์ชันคลื่นเหล่านี้เป็นเวกเตอร์ของแอมพลิจูดเชิงซ้อน โดยแต่ละแอมพลิจูดสอดคล้องกับการมีส่วนร่วมของการจัดวางอิเล็กตรอนที่เป็นไปได้
ground state คือฟังก์ชันคลื่นที่มีพลังงานต่ำสุดของระบบ และมีความสำคัญพิเศษในการศึกษาระบบโมเลกุล วิธีการที่แม่นยำที่สุดในการคำนวณ ground state พิจารณาการจัดวางอิเล็กตรอนทั้งหมดที่เป็นไปได้ แต่วิธีนี้ไม่สามารถทำได้จริงสำหรับระบบขนาดใหญ่ เนื่องจากจำนวนการจัดวางเพิ่มขึ้นแบบ exponential ตามขนาดของระบบ
Handover Iterative Variational Quantum Eigensolver (HI-VQE) เป็นวิธีไฮบริดควอนตัม-คลาสสิกที่ล้ำสมัยสำหรับการประมาณ ground state ของระบบโมเลกุลอย่างแม่นยำ ผสานรวม quantum hardware กับการคำนวณแบบคลาสสิก โดยใช้ quantum processor เพื่อสำรวจการจัดวางอิเล็กตรอนที่เป็นผู้สมัครอย่างมีประสิทธิภาพ และคำนวณฟังก์ชันคลื่นผลลัพธ์บนคอมพิวเตอร์คลาสสิก ด้วยการสร้างฟังก์ชันคลื่นที่กระชับแต่มีความแม่นยำทางเคมี HI-VQE ช่วยส่งเสริมการวิจัยและการค้นพบในเคมีควอนตัมและวิทยาศาสตร์วัสดุ

HI-VQE ลดความซับซ้อนทางการคำนวณของปัญหาโครงสร้างอิเล็กตรอนด้วยการประมาณ ground state อย่างมีประสิทธิภาพด้วยความแม่นยำสูง มุ่งเน้นที่ชุดย่อยของการจัดวางอิเล็กตรอนที่เกี่ยวข้องมากที่สุดที่คัดเลือกมาอย่างดี เพื่อเพิ่มประสิทธิภาพทั้งความแม่นยำและประสิทธิภาพการคำนวณ
ด้วยการรวมจุดแข็งของทั้งคอมพิวเตอร์คลาสสิกและควอนตัม HI-VQE ปรับปรุงและพัฒนาการประมาณฟังก์ชันคลื่นปัจจุบันแบบ iterative เทคนิคการสร้าง subspace เฉพาะตัวช่วยให้การเลือกการจัดวางมีประสิทธิภาพมากขึ้น ทำให้ผู้ใช้มีการควบคุมการคำนวณที่มากขึ้นและความแม่นยำที่ดีขึ้นในการจำลองเคมีควอนตัม
หากต้องการเรียนรู้เกี่ยวกับอัลกอริทึมในเชิงลึกมากขึ้น สามารถ อ่านบทความวิจัยที่เกี่ยวข้อง ได้
คำอธิบาย
จำนวนการจัดวางอิเล็กตรอนสำหรับระบบโมเลกุลเพิ่มขึ้นแบบ exponential ตามขนาดระบบ อย่างไรก็ตาม สำหรับสถานะอิเล็กทรอนิกส์บางอย่าง เช่น ground state มักพบว่ามีเพียงส่วนเล็กน้อยของการจัดวางที่มีส่วนร่วมอย่างมีนัยสำคัญต่อพลังงานของสถานะนั้น วิธี Selected configuration interaction (SCI) ใช้ประโยชน์จากความกระจัดกระจายนี้เพื่อลดต้นทุนการคำนวณโดยการระบุและมุ่งเน้นที่การจัดวางที่เกี่ยวข้องมากที่สุด ชุดย่อยของการจัดวางนี้เรียกว่า subspace
HI-VQE ใช้ประสิทธิภาพโดยธรรมชาติของ quantum computer ในการแสดงแทนระบบโมเลกุลเพื่อช่วยในการค้นหา subspace ผสมผสาน subroutine คลาสสิกและควอนตัมเพื่อแก้ปัญหาโครงสร้างอิเล็กตรอนด้วยความแม่นยำสูง ต่างจากวิธี quantum SCI ที่มีอยู่ HI-VQE รวมการฝึก variational, การสร้าง subspace แบบ iterative และการคัดกรองการจัดวางแบบ pre-diagonalization เพื่อเพิ่มประสิทธิภาพด้วยการลดการวัดควอนตัม, การ iteration และต้นทุน diagonalization แบบคลาสสิก HI-VQE จึงสามารถนำไปใช้กับระบบโมเลกุลขนาดใหญ่ขึ้นที่ต้องการ Qubit มากขึ้น และลดต้นทุนในการแก้ปัญหาขนาดที่กำหนดด้วยระดับความแม่นยำเดิม
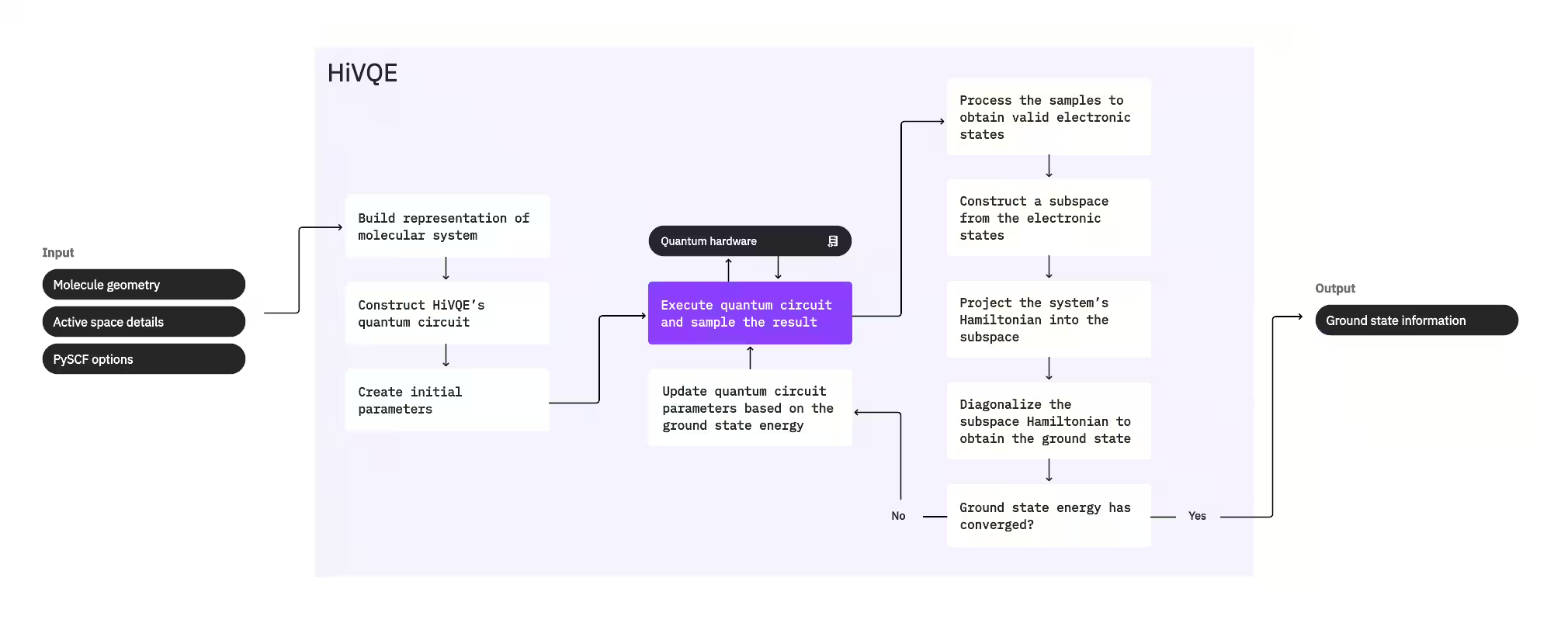
ในการคำนวณ ground state ของระบบ HI-VQE จะใช้แพ็กเกจเคมี PySCF แบบคลาสสิกก่อนเพื่อสร้างการแสดงแทนโมเลกุลจาก input ที่ผู้ใช้ระบุ เช่น ข้อมูลทางเรขาคณิตของโมเลกุลและข้อมูลโมเลกุลอื่น ๆ จากนั้นเข้าสู่ loop การ optimize แบบไฮบริดควอนตัม-คลาสสิก โดย iterative ปรับปรุง subspace เพื่อแสดงแทน ground state ได้อย่างเหมาะสมในขณะที่ลดจำนวนการจัดวางให้น้อยที่สุด loop จะดำเนินต่อไปจนกว่าจะตรงตามเกณฑ์การบรรจบ เช่น ขนาด subspace หรือความเสถียรของพลังงาน จากนั้นจึงส่งออกฟังก์ชันคลื่น ground state และพลังงานที่คำนวณได้ ผลลัพธ์เหล่านี้สามารถใช้สร้าง potential energy surface ที่แม่นยำและทำการวิเคราะห์เพิ่มเติมของระบบ
loop การ optimize มุ่งเน้นที่การปรับ parameter ของ quantum circuit เพื่อสร้าง subspace คุณภาพสูง HI-VQE มีตัวเลือก quantum circuit สามแบบ: excitation_preserving, efficient_su2 และ LUCJ การ optimize เริ่มต้นใกล้กับ reference state ของ Hartree-Fock เนื่องจากมีความเหมาะสมทั่วไป จากนั้น Circuit จะถูกประมวลผลบน quantum device และการจัดวางจะถูก sample จาก quantum state ผลลัพธ์ก่อนส่งกลับมาเป็น binary string เนื่องจาก noise ของ quantum device บางการจัดวางที่ sample มาอาจไม่ถูกต้องทางกายภาพ ล้มเหลวในการอนุรักษ์จำนวนอิเล็กตรอนหรือ spin HI-VQE แก้ปัญหานี้โดยใช้กระบวนการ configuration recovery จากแพ็กเกจ qiskit-addon-sqd เพื่อให้ผู้ใช้สามารถแก้ไขการจัดวางที่ไม่ถูกต้องหรือละทิ้งได้
การจัดวางที่ถูกต้องจะผ่านขั้นตอนการคัดกรองเพิ่มเติมโดยเป็นทางเลือก เพื่อลบรายการที่คาดว่าจะมีส่วนร่วมน้อยที่สุด ซึ่งลดขนาดของ subspace ทำให้ลดต้นทุนของขั้นตอน diagonalization หากเปิดใช้งานการคัดกรอง จะสร้าง subspace Hamiltonian เบื้องต้นจากการจัดวางที่ถูกต้อง และทำ diagonalization ด้วยเกณฑ์การสิ้นสุดที่ผ่อนปรนมาก แม้ว่าความแม่นยำของ amplitude ผลลัพธ์สำหรับแต่ละการจัดวางจะต่ำ แต่มีประสิทธิภาพในการทำนายว่าการจัดวางใดควรถูกยกเว้นจาก subspace ใน iteration นี้ และคำนวณได้รวดเร็ว
การจัดวางที่เลือกจะถูกเพิ่มเข้าใน subspace และ Hamiltonian ของระบบจะถูก project เข้าสู่ subspace นี้ subspace อัปเดตแบบ iterative โดยเก็บรักษาการจัดวางที่เกี่ยวข้องมากที่สุดในแต่ละ iteration แนวทางนี้แตกต่างจากวิธีอื่นเพราะ quantum circuit ไม่จำเป็นต้องประมาณ ground state เต็มรูปแบบในแต่ละขั้นตอน
ต่อจากนั้น subspace Hamiltonian จะถูก diagonalize แบบคลาสสิกเพื่อหา eigenvalue ต่ำสุดและ eigenvector ที่สอดคล้อง ซึ่งแสดงการประมาณ ground state และพลังงานของมัน เมื่อคุณภาพ subspace ดีขึ้นผ่านการ iteration ground state ที่คำนวณได้จะประมาณ ground state จริงได้ดีขึ้น ขั้นตอนการคัดกรองเพิ่มเติมสามารถดำเนินการได้ ณ จุดนี้ เพื่อลบการจัดวางที่ไม่มีส่วนร่วมอย่างมีนัยสำคัญใน ground state ที่คำนวณได้ออกจาก subspace ขั้นตอนนี้ทำให้ subspace ที่นำเข้าสู่ iteration ถัดไปกระชับที่สุด โดยประเมินจาก amplitude ที่ได้จาก diagonalization เนื่องจากสิ่งเหล่านี้แสดงถึงความสำคัญของการมีส่วนร่วมของแต่ละการจัดวางต่อ ground state ที่คำนวณได้
การตรวจสอบการบรรจบจะกำหนดว่าการฝึกเพิ่มเติมจะปรับปรุงผลลัพธ์หรือไม่ ถ้าใช่ จะทำขั้นตอน classical expansion เพิ่มเติม parameter ของ quantum circuit จะถูกอัปเดตเพื่อลดพลังงานที่คำนวณได้เพิ่มเติม และกระบวนการจะวนซ้ำ ขั้นตอน classical expansion สร้างการจัดวางเพิ่มเติมสำหรับ subspace โดย supplement การจัดวางที่ sample จาก quantum device โดยระบุการจัดวางที่มี amplitude ใหญ่สุดในผลการ diagonalization ก่อน จากนั้นสร้างการจัดวางใหม่ด้วย single และ double excitation จากการจัดวางที่ระบุ แล้วนำการจัดวางตามจำนวนที่ต้องการเพิ่มเข้าใน subspace
เมื่อพิจารณาแล้วว่า iteration บรรจบแล้ว HI-VQE จะส่งคืน ground state ที่คำนวณได้ (ในรูปแบบของสถานะใน subspace และ amplitude ของสถานะเหล่านั้นในฟังก์ชันคลื่น ground state), พลังงานของมัน และค่าวัด energy variance ที่บอกว่าสถานะที่คำนวณได้เป็น eigenstate ของ Hamiltonian ของระบบหรือไม่
ผู้ใช้สามารถกำหนด quantum circuit ที่ใช้และจำนวน shot สำหรับแต่ละ quantum circuit รวมถึงควบคุมขนาด subspace หรือเปิดใช้งานการสร้างการจัดวางเพิ่มเติมแบบคลาสสิกเพื่อช่วย quantum generated configuration ในลักษณะนี้ผู้ใช้สามารถปรับพฤติกรรมของ HI-VQE ให้เหมาะสมกับแอปพลิเคชันที่ต้องการได้
การอนุญาตสิทธิ์
โปรดทราบว่าการใช้งาน Qiskit Function นี้จำกัดเฉพาะปัญหาที่ต้องใช้ Qubit ไม่เกิน 20 ตัว เว้นแต่จะได้รับใบอนุญาตที่อนุญาตให้ใช้จำนวนสูงกว่า
หากต้องการสอบถามเกี่ยวกับการขอใบอนุญาต สามารถส่งอีเมลไปที่ qiskit.support@qunovacomputing.com
เริ่มต้นใช้งาน
ขั้นแรก ขอสิทธิ์เข้าถึง function จากนั้น authenticate โดยใช้ IBM Quantum® API key และสมมติว่าคุณได้ บันทึกบัญชีของคุณ ในสภาพแวดล้อมในเครื่องแล้ว เลือก Qiskit Function ดังนี้:
import reprlib
from qiskit_ibm_catalog import QiskitFunctionsCatalog
catalog = QiskitFunctionsCatalog(channel="ibm_quantum_platform")
function = catalog.load("qunova/hivqe-chemistry")
ตัวอย่าง
ตัวอย่างแรกแสดงวิธีคำนวณพลังงาน ground state สำหรับโมเลกุล NH3 โดยใช้อัลกอริทึม HI-VQE
กำหนดเรขาคณิตของโมเลกุลและตัวเลือก
เรขาคณิตของโมเลกุล NH3 ระบุด้วยพิกัด Cartesian ที่คั่นด้วย ";" สำหรับแต่ละอะตอม
# Define the molecule geometry
geometry = """
N -0.85188 -0.02741 0.03141;
H 0.16545 0.00593 -0.01648;
H -1.16348 -0.39357 -0.86702;
H -1.16348 0.94228 0.06281;
"""
สามารถกำหนดและระบุตัวเลือกเพิ่มเติมสำหรับระบบโมเลกุลในรูปแบบ dictionary ต่อไปนี้
# Configure some options for the job.
molecule_options = {"basis": "sto3g"}
hivqe_options = {"shots": 100, "max_iter": 20}
ประมวลผล function ด้วย input เรขาคณิตและตัวเลือก
# Run HI-VQE
job = function.run(
geometry=geometry,
# `backend_name` is the name of a backend with at least 16 qubits,
# for example, "ibm_marrakesh".
backend_name=backend_name,
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
ควร print Function job ID เพื่อให้สามารถใส่ใน support request ได้หากมีปัญหา
print("Job ID:", job.job_id)
Job ID: e5ced6f2-fd1d-4244-a6aa-bd27cfb0cdee
ตัวอย่างนี้ใช้ 16 Qubit กับ 8 orbital ของ basis sto3g สำหรับโมเลกุล NH3 ตรวจสอบสถานะของ Qiskit Function workload หรือดึงผลลัพธ์ดังนี้:
print(job.status())
QUEUED
หลังจาก job เสร็จสิ้น สามารถรับผลลัพธ์ด้วย instance result()
result = job.result()
# Output can be long, so we display a shortened representation
shortened_result = reprlib.repr(result)
print(shortened_result)
{'eigenvector': [0.9824448589364075, 0.009527106392132133, 6.854074372058527e-08, 3.591500190038039e-07, 0.0012975231577544268, 2.310159709002111e-05, ...], 'energy': -55.52108557170985, 'energy_history': [-55.51901898989887, -55.52056881448526, -55.52065046778772, -55.520690696813716, -55.520691108428, -55.520708448092634, ...], 'energy_variance': 3.066239097617371e-10, ...}
ในการเข้าถึงพลังงาน ground state ให้ใช้ key "energy" key "eigenvector" ให้ CI coefficient พร้อมกับสัญลักษณ์ bitstring ของการจัดวางอิเล็กตรอนที่เก็บไว้ใน "states" ของผลลัพธ์
fci_energy = -55.521148034704126 # the exact energy using FCI method
hivqe_energy = result["energy"]
print(
f"|Exact Energy - HI-VQE Energy|: "
f"{abs(fci_energy - hivqe_energy) * 1000} mHa"
)
print(f"Sampled Number of States: {len(result['states'])}")
|Exact Energy - HI-VQE Energy|: 0.06246299427914437 mHa
Sampled Number of States: 1936
ประสิทธิภาพ
ส่วนนี้แสดงการคำนวณ benchmark ที่พิสูจน์แล้วของ HI-VQE กับ 24 Qubit สำหรับ Li2S, 40 Qubit สำหรับโมเลกุล N2 และ 44 Qubit สำหรับระบบ FeP-NO
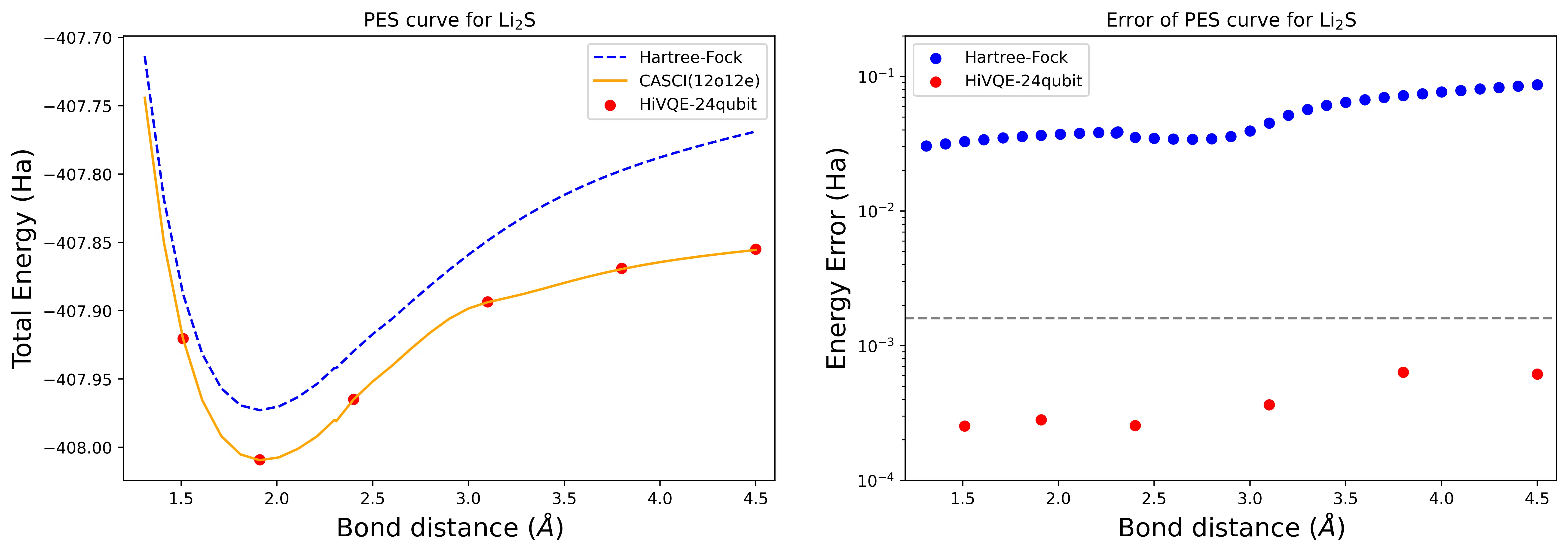
Dissociation potential energy surface curve สำหรับโมเลกุล Li2S ด้วย 24 Qubit
แสดง PES curve พร้อม FCI reference และ initial guess จาก RHF รวมถึง energy error จาก FCI reference
การคำนวณดำเนินการด้วยเรขาคณิตและตัวเลือกต่อไปนี้
# This cell is hidden from users
backend_name = service.least_busy(operational=True, min_num_qubits=38).name
# Define Li2S geometries
Li2S_geoms = {
"Li2S_1.51": "S -1.239044 0.671232 -0.030374;Li -1.506327 0.432403 -1.498949;Li -0.899996 0.973348 1.826768;",
"Li2S_2.40": "S -1.741432 0.680397 0.346702;Li -0.529307 0.488006 -1.729343;Li -1.284307 0.989409 2.177209;",
"Li2S_3.80": "S -2.707255 0.674298 0.909161;Li 0.079218 0.552012 -1.671656;Li -0.927010 0.931502 1.557063;",
}
# Configure some options for the job.
molecule_options = {
"basis": "sto3g",
}
hivqe_options = {
"shots": 100,
"max_iter": 20,
}
results = []
for geom in ["Li2S_1.51", "Li2S_2.40", "Li2S_3.80"]:
# Run HI-VQE
job = function.run(
geometry=Li2S_geoms[geom],
backend_name=backend_name, # can use any device with at least 38 qubits
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
results.append(job.result())
จุดสีแดงแสดงผลการคำนวณ HI-VQE สำหรับหกเรขาคณิตที่แตกต่างกัน และ input ในเซลล์ข้างต้นระบุสามเรขาคณิตที่สอดคล้องกับ 1.51, 2.40 และ 3.80 Angstrom
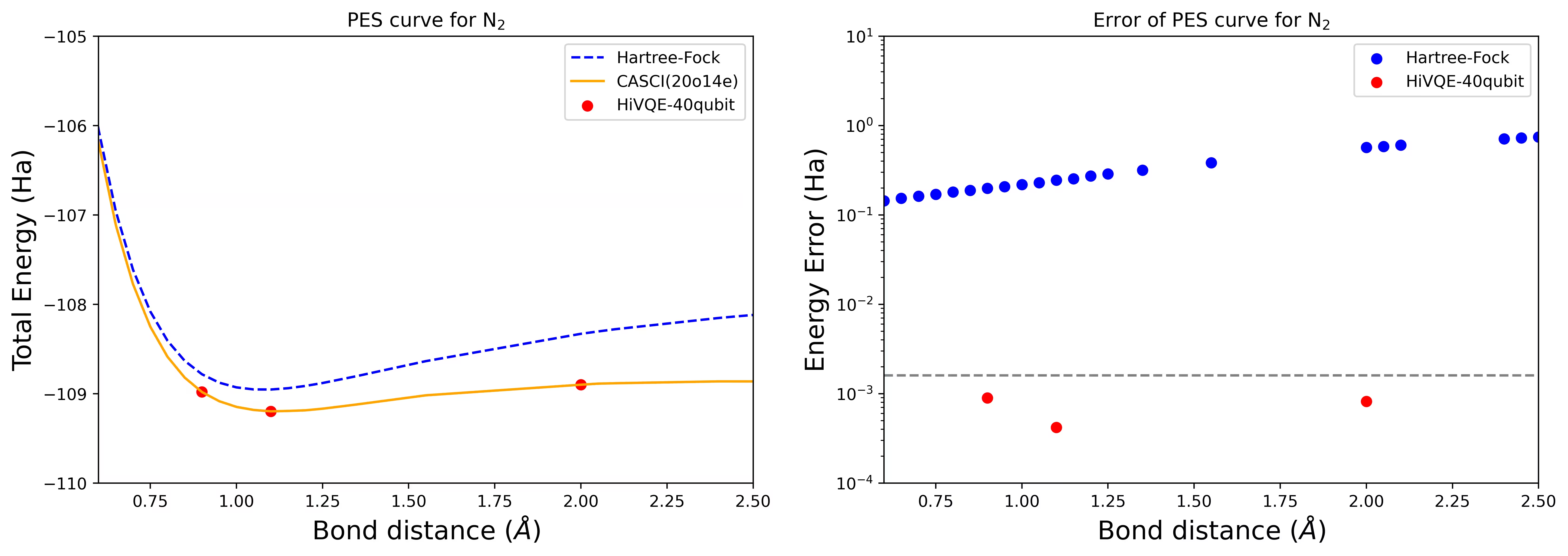
Dissociation PES curve สำหรับโมเลกุล N2 ด้วย 40 Qubit
โมเลกุลไนโตรเจนได้รับการระบุว่าเป็นระบบ multireference ที่มีการมีส่วนร่วมของพลังงาน correlation ขนาดใหญ่นอกเหนือจาก Hartree-Fock state เราดำเนินการคำนวณ benchmark สำหรับโมเลกุล N2 ด้วย basis cc-pvdz, (20o,14e) โดยใช้การเลือก active orbital แบบ homo-lumo จำนวน complete active space (CAS) เพื่อแสดงแทนปัญหานี้คือ 6,009,350,400 ไม่สามารถหาคำตอบ eigenvalue problem (สำหรับพลังงานและโครงสร้างอิเล็กทรอนิกส์) ด้วยสถานะจำนวนนี้โดยใช้เดสก์ท็อปที่ทรงพลัง (16cpu/64GB) ด้วย HI-VQE ผู้ใช้สามารถค้นหา subspace ของ CAS state อย่างมีประสิทธิภาพเพื่อหาผลลัพธ์ที่มีความแม่นยำทางเคมีในขณะที่ประหยัดทรัพยากรการคำนวณอย่างมีนัยสำคัญ กราฟต่อไปนี้แสดง PES curve ของการคำนวณ HI-VQE 40 Qubit สำหรับ dissociation ของโมเลกุล N2
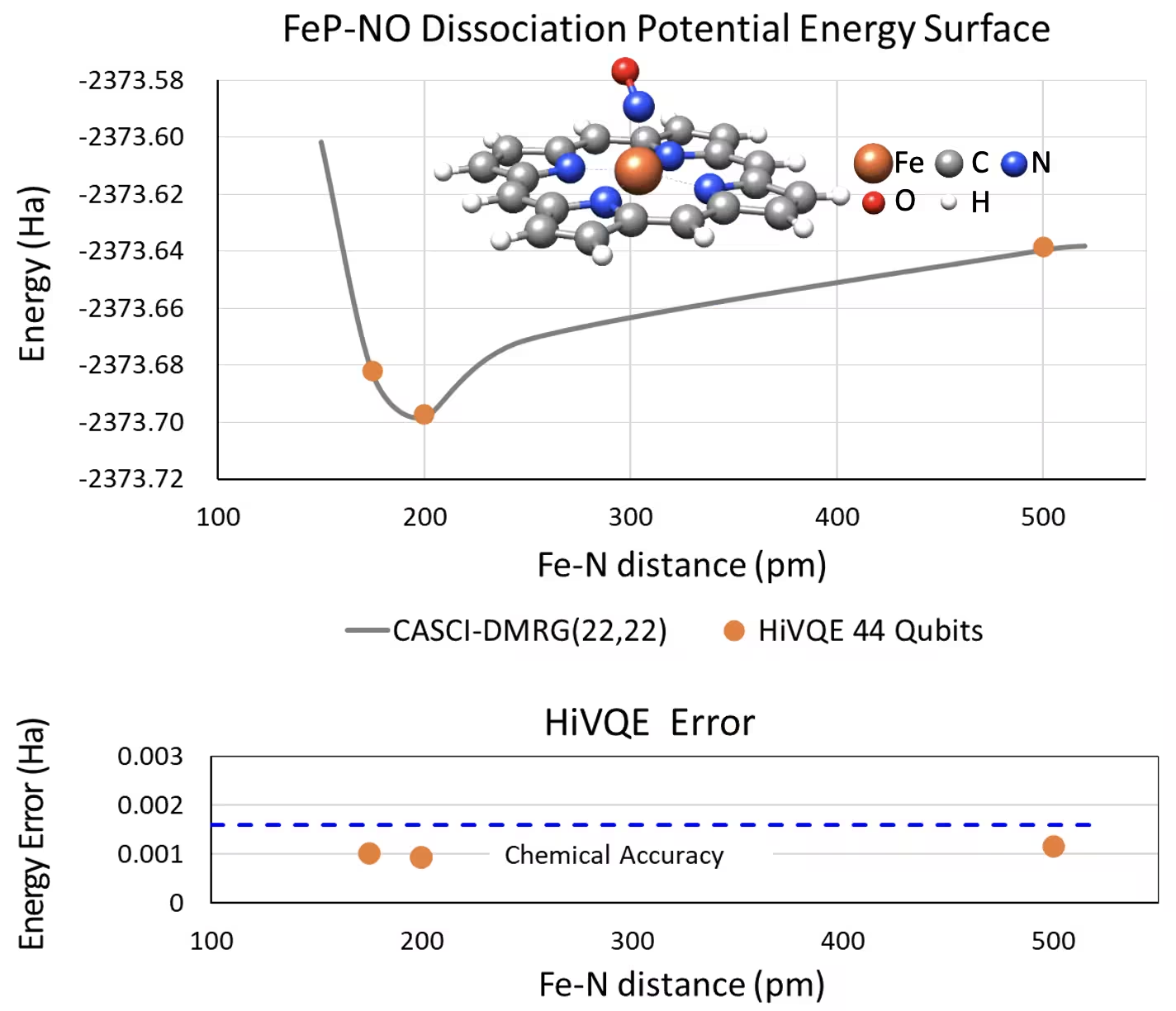
Dissociation PES curve สำหรับ five-coordinated iron(II)-porphyrin กับระบบ NO ด้วย 44 Qubit
ระบบเคมีที่น่าสนใจอีกอย่างคือ iron(II)-porphyrin (FeP) complex ที่มี nitric oxide (NO) ligand ประสาน ซึ่งเป็นระบบ metalloporphyrin ที่เกี่ยวข้องทางชีววิทยาซึ่งมีบทบาทสำคัญในกระบวนการทางสรีรวิทยาต่าง ๆ ในตัวอย่างนี้ HI-VQE ถูกใช้เพื่อประมาณ potential energy surface curve ที่แม่นยำของปฏิกิริยา intermolecular ระหว่าง FeP และ NO (พลังงาน ground state สำหรับเรขาคณิตที่แยกกันต่างกัน) ระบบรวมมี 450 orbital และ 202 อิเล็กตรอน (450o,202e) ด้วย basis 6-31g(d) รวมทั้งหมด การเลือก active orbital แบบ homo-lumo ถูกใช้เพื่อคำนวณกรณีที่เล็กกว่าจากกรณีจริงด้วย (22o,22e) จากผลลัพธ์ benchmark ต่อไปนี้ เราสามารถบรรลุความแม่นยำทางเคมี (> 1.6 mHa) กับการคำนวณเคมีคอมพิวเตอร์คลาสสิกล้ำสมัยของ CASCI(DMRG) (22o,22e) reference
Benchmark
- Exact matrix size คือจำนวน determinant สำหรับคำตอบที่แน่นอน เช่น FCI และ CASCI
- การคำนวณ HI-VQE sample และคำนวณ subspace ของมัน (นั่นคือ HI-VQE matrix size)
- Total time รวม QPU runtime และ Qiskit Function ที่รันด้วย CPU
- ความแม่นยำประมาณจาก energy difference จากคำตอบที่แน่นอน
| ระบบเคมี | จำนวน Qubit | Exact matrix size | HI-VQE matrix size | E(diff) จากที่แน่นอน (mHa) | จำนวน iteration | เวลารวม | QPU runtime usage |
|---|---|---|---|---|---|---|---|
| (8o,10e) | 16 | 3136 | 1936 | 0.08 | 6 | 37 s | 34 s |
| (10o,10e) | 20 | 63504 | 3969 | 0.60 | 5 | 250 s | 50 s |
| (15o,10e) | 30 | 9018009 | 49729 | 0.90 | 5 | 354 s | 54 s |
| (16o,14e) | 32 | 130873600 | 1798281 | 1.10 | 9 | 6531 s | 121 s |
| (18o,24e) | 36 | 344622096 | 399424 | 0.90 | 24 | 5174 s | 130 s |
| (20o,14e) | 40 | 6009350400 | 9012004 | 1.20 | 21 | 46547 s | 258 s |
ดึง error message
หาก workload ล้มเหลว สถานะจะเป็น ERROR และการเรียก job.result() จะเกิด exception:
job = function.run(
geometry="invalid-geometry", # This will cause an error
backend_name=backend_name,
max_states=2000,
max_expansion_states=15,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
job.result()
job.status()
'ERROR'
รับการสนับสนุน
สามารถส่งอีเมลไปที่ qiskit.support@qunovacomputing.com สำหรับความช่วยเหลือเกี่ยวกับ function นี้
หากต้องการความช่วยเหลือในการแก้ไขปัญหา error เฉพาะ โปรดระบุ Function job ID ของ job ที่พบ error ด้วย
ขั้นตอนถัดไป
- ขอสิทธิ์เข้าถึง function โดยกรอกแบบฟอร์มนี้
- ดู API reference สำหรับ Qiskit Function นี้
- ลองทำ tutorial Compute dissociation PES curve for FeP-NO with HI-VQE
- อ่าน Pellow-Jarman, A., et al. (2025). HIVQE: Handover Iterative Variational Quantum Eigensolver for Efficient Quantum Chemistry Calculations. arXiv preprint arXiv:2503.06292.
- ลองทำ tutorial Dissociation PES curves with Qunova HiVQE