การวิเคราะห์เมทริกซ์เชิงควอนตัมแบบ Krylov โดยใช้การสุ่มตัวอย่างสำหรับโมเดลแลตทิซเฟอร์มิออน
ประมาณการการใช้งาน: เก้าวินาทีบนโปรเซสเซอร์ Heron r2 (หมายเหตุ: นี่เป็นเพียงการประมาณการเท่านั้น เวลาจริงของคุณอาจแตกต่างออกไป)
ผลลัพธ์การเรียนรู้
หลังจากผ่านบทแนะนำนี้แล้ว ผู้ใช้ควรเข้าใจ:
- วิธีใช้ SQD Qiskit addon เพื่อประมาณ ground state energy ของโมเดลแลตทิซโดยใช้ bitstring ที่สุ่มตัวอย่างจากหน่วยประมวลผลควอนตัม (QPU)
- วิธีใช้ ffsim เพื่อสร้าง time evolution circuit สำหรับการจำลองเฟอร์มิออน
- วิธีรวม sample จาก Circuit หลายตัวสำหรับการ post-process ด้วยอัลกอริทึม sample-based Krylov diagonalization (SKQD)
ข้อกำหนดเบื้องต้น
เราแนะนำให้ผู้ใช้คุ้นเคยกับหัวข้อต่อไปนี้ก่อนเรียนบทแนะนำนี้:
- Sample-based quantum diagonalization of a chemistry Hamiltonian
- Krylov quantum diagonalization of lattice Hamiltonians
- Qiskit primitives
พื้นหลัง
บทแนะนำนี้แสดงวิธีใช้ sample-based quantum diagonalization (SQD) เพื่อประมาณพลังงานสถานะพื้นฐานของโมเดลแลตทิซเฟอร์มิออน โดยเฉพาะอย่างยิ่ง เราศึกษา one-dimensional single-impurity Anderson model (SIAM) ซึ่งใช้อธิบายสิ่งเจือปนแม่เหล็กที่ฝังอยู่ในโลหะ
บทแนะนำนี้ทำตามขั้นตอนการทำงานที่คล้ายกับบทแนะนำที่เกี่ยวข้อง Sample-based quantum diagonalization of a chemistry Hamiltonian แต่ความแตกต่างหลักอยู่ที่วิธีการสร้าง Circuit ควอนตัม บทแนะนำอื่นใช้ heuristic variational ansatz ซึ่งน่าสนใจสำหรับ Hamiltonian ทางเคมีที่อาจมีพจน์ปฏิสัมพันธ์หลายล้านพจน์ ในทางกลับกัน บทแนะนำนี้ใช้ Circuit ที่ประมาณการวิวัฒนาการตามเวลาด้วย Hamiltonian Circuit เหล่านี้อาจมีความลึกมาก ซึ่งทำให้วิธีนี้เหมาะสมกว่าสำหรับการประยุกต์ใช้กับโมเดลแลตทิซ เวกเตอร์สถานะที่ Circuit เหล่านี้สร้างขึ้นก่อให้เกิดฐานสำหรับ Krylov subspace และด้วยเหตุนี้ อัลกอริทึมจึงสามารถพิสูจน์ได้และลู่เข้าสู่สถานะพื้นฐานอย่างมีประสิทธิภาพ ภายใต้ข้อสมมติฐานที่เหมาะสม
วิธีการที่ใช้ในบทแนะนำนี้สามารถมองได้ว่าเป็นการผสมผสานเทคนิคที่ใช้ใน SQD และ Krylov quantum diagonalization (KQD) วิธีการรวมนี้บางครั้งเรียกว่า sample-based Krylov quantum diagonalization (SQKD) ดู Krylov quantum diagonalization of lattice Hamiltonians สำหรับบทแนะนำเกี่ยวกับวิธี KQD
บทแนะนำนี้อ้างอิงจากงานวิจัย "Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization" ซึ่งสามารถอ้างถึงเพื่อดูรายละเอียดเพิ่มเติมได้
Single-impurity Anderson model (SIAM)
SIAM Hamiltonian หนึ่งมิติเป็นผลรวมของสามพจน์:
โดยที่
ที่นี่ คือตัวดำเนินการสร้าง/ทำลายเฟอร์มิออนสำหรับตำแหน่ง bath ที่ ที่มีสปิน , คือตัวดำเนินการสร้าง/ทำลายสำหรับโหมดสิ่งเจือปน และ ส่วน , และ เป็นจำนวนจริงที่อธิบายปฏิสัมพันธ์การกระโดด on-site และ hybridization และ เป็นจำนวนจริงที่ระบุศักย์เคมี
โปรดสังเกตว่า Hamiltonian นี้เป็นกรณีเฉพาะของ Hamiltonian อิเล็กตรอนปฏิสัมพันธ์ทั่วไป
โดยที่ ประกอบด้วยพจน์หนึ่งอนุภาค ซึ่งเป็นกำลังสองในตัวดำเนินการสร้างและทำลายเฟอร์มิออน และ ประกอบด้วยพจน์สองอนุภาค ซึ่งเป็นกำลังสี่ สำหรับ SIAM:
และ มีพจน์ที่เหลือใน Hamiltonian เพื่อแทน Hamiltonian ด้วยโปรแกรม เราเก็บเมทริกซ์ และเทนเซอร์
ฐานตำแหน่งและฐานโมเมนตัม
เนื่องจากความสมมาตรการเลื่อนตำแหน่งโดยประมาณใน เราจึงไม่คาดว่าสถานะพื้นฐานจะกระจายตัวน้อยในฐานตำแหน่ง (ฐานออร์บิทัลที่ระบุ Hamiltonian ข้างต้น) ประสิทธิภาพของ SQD รับประกันได้เฉพาะเมื่อสถานะพื้นฐานกระจายตัวน้อย นั่นคือมีน้ำหนักสำคัญบนสถานะฐานการคำนวณเพียงไม่กี่สถานะ เพื่อปรับปรุงความกระจัดกระจายของสถานะพื้นฐาน เราทำการจำลองในฐานออร์บิทัลที่ เป็นแนวทแยง เราเรียกฐานนี้ว่า ฐานโมเมนตัม เนื่องจาก เป็น Hamiltonian เฟอร์มิออนกำลังสอง จึงสามารถทำให้เป็นแนวทแยงได้อย่างมีประสิทธิภาพโดยการหมุนออร์บิทัล
การประมาณวิวัฒนาการตามเวลาด้วย Hamiltonian
เพื่อประมาณวิวัฒนาการตามเวลาด้วย Hamiltonian เราใช้การแตกตัวแบบ second order Trotter-Suzuki:
ภายใต้ Jordan-Wigner transformation วิวัฒนาการตามเวลาด้วย เท่ากับ Gate CPhase เดียวระหว่างออร์บิทัล spin-up และ spin-down ที่ตำแหน่งสิ่งเจือปน เนื่องจาก เป็น Hamiltonian เฟอร์มิออนกำลังสอง วิวัฒนาการตามเวลาด้วย จึงเท่ากับการหมุนออร์บิทัล
สถานะฐาน Krylov โดยที่ คือมิติของ Krylov subspace ถูกสร้างขึ้นโดยการใช้ขั้นตอน Trotter ซ้ำ ดังนั้น
ในขั้นตอนการทำงานแบบ SQD ต่อไปนี้ เราจะสุ่มตัวอย่างจากชุด Circuit นี้และประมวลผลชุด bitstring รวมกันด้วย SQD วิธีนี้แตกต่างจากที่ใช้ในบทแนะนำที่เกี่ยวข้อง Sample-based quantum diagonalization of a chemistry Hamiltonian ซึ่งสุ่มตัวอย่างจาก Circuit variational เชิงฮิวริสติกเดียว
ข้อกำหนด
ก่อนเริ่มบทแนะนำนี้ ตรวจสอบให้แน่ใจว่าติดตั้งสิ่งต่อไปนี้แล้ว:
- Qiskit SDK v1.0 หรือใหม่กว่า พร้อมรองรับ visualization
- Qiskit Runtime v0.22 หรือใหม่กว่า (
pip install qiskit-ibm-runtime) - SQD Qiskit addon v0.11 หรือใหม่กว่า (
pip install qiskit-addon-sqd) - ffsim v0.0.72 หรือใหม่กว่า (
pip install ffsim)
ตัวอย่างขนาดเล็กด้วยซิมูเลเตอร์
ขั้นตอนที่ 1: แมปปัญหากับ Circuit ควอนตัม
ก่อนอื่น เราสร้าง SIAM Hamiltonian ในฐานตำแหน่ง โดย Hamiltonian แทนด้วยเมทริกซ์ และเทนเซอร์ จากนั้นเราหมุนไปยังฐานโมเมนตัม ในฐานตำแหน่ง เราวางสิ่งเจือปนไว้ที่ตำแหน่งแรก แต่เมื่อหมุนไปยังฐานโมเมนตัม เราย้ายสิ่งเจือปนไปยังตำแหน่งกลางเพื่อให้ปฏิสัมพันธ์กับออร์บิทัลอื่นได้ง่ายขึ้น
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime scipy
import numpy as np
import pyscf.fci
def siam_hamiltonian(
norb: int,
hopping: float,
onsite: float,
hybridization: float,
chemical_potential: float,
) -> tuple[np.ndarray, np.ndarray]:
"""Hamiltonian for the single-impurity Anderson model."""
# Place the impurity on the first site
impurity_orb = 0
# One body matrix elements in the "position" basis
h1e = np.zeros((norb, norb))
np.fill_diagonal(h1e[:, 1:], -hopping)
np.fill_diagonal(h1e[1:, :], -hopping)
h1e[impurity_orb, impurity_orb + 1] = -hybridization
h1e[impurity_orb + 1, impurity_orb] = -hybridization
h1e[impurity_orb, impurity_orb] = chemical_potential
# Two body matrix elements in the "position" basis
h2e = np.zeros((norb, norb, norb, norb))
h2e[impurity_orb, impurity_orb, impurity_orb, impurity_orb] = onsite
return h1e, h2e
def momentum_basis(norb: int) -> np.ndarray:
"""Get the orbital rotation to change from the position to the momentum basis."""
n_bath = norb - 1
# Orbital rotation that diagonalizes the bath (non-interacting system)
hopping_matrix = np.zeros((n_bath, n_bath))
np.fill_diagonal(hopping_matrix[:, 1:], -1)
np.fill_diagonal(hopping_matrix[1:, :], -1)
_, vecs = np.linalg.eigh(hopping_matrix)
# Expand to include impurity
orbital_rotation = np.zeros((norb, norb))
# Impurity is on the first site
orbital_rotation[0, 0] = 1
orbital_rotation[1:, 1:] = vecs
# Move the impurity to the center
new_index = n_bath // 2
perm = np.r_[1 : (new_index + 1), 0, (new_index + 1) : norb]
orbital_rotation = orbital_rotation[:, perm]
return orbital_rotation
def rotated(
h1e: np.ndarray, h2e: np.ndarray, orbital_rotation: np.ndarray
) -> tuple[np.ndarray, np.ndarray]:
"""Rotate the orbital basis of a Hamiltonian."""
h1e_rotated = np.einsum(
"ab,Aa,Bb->AB",
h1e,
orbital_rotation,
orbital_rotation.conj(),
optimize="greedy",
)
h2e_rotated = np.einsum(
"abcd,Aa,Bb,Cc,Dd->ABCD",
h2e,
orbital_rotation,
orbital_rotation.conj(),
orbital_rotation,
orbital_rotation.conj(),
optimize="greedy",
)
return h1e_rotated, h2e_rotated
# Total number of spatial orbitals, including the bath sites and the impurity
# This should be an even number
norb = 8
# System is half-filled
nelec = (norb // 2, norb // 2)
# One orbital is the impurity, the rest are bath sites
n_bath = norb - 1
# Hamiltonian parameters
hybridization = 1.0
hopping = 1.0
onsite = 10.0
chemical_potential = -0.5 * onsite
# Generate Hamiltonian in position basis
h1e, h2e = siam_hamiltonian(
norb=norb,
hopping=hopping,
onsite=onsite,
hybridization=hybridization,
chemical_potential=chemical_potential,
)
# Rotate to momentum basis
orbital_rotation = momentum_basis(norb)
h1e_momentum, h2e_momentum = rotated(h1e, h2e, orbital_rotation.T.conj())
# In the momentum basis, the impurity is placed in the center
impurity_index = n_bath // 2
# Use PySCF to compute the exact ground state energy
reference_energy, _ = pyscf.fci.direct_spin1.kernel(h1e, h2e, norb, nelec)
from typing import Sequence
import ffsim
import scipy
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.circuit import CircuitInstruction, Qubit
from qiskit.circuit.library import CPhaseGate, XGate, XXPlusYYGate
def prepare_initial_state(qubits: Sequence[Qubit], norb: int, nocc: int):
"""Prepare initial state."""
assert norb >= 8
x_gate = XGate()
rot = XXPlusYYGate(0.5 * np.pi, -0.5 * np.pi)
for i in range(nocc):
yield CircuitInstruction(x_gate, [qubits[i]])
yield CircuitInstruction(x_gate, [qubits[norb + i]])
for i in range(3):
for j in range(nocc - i - 1, nocc + i, 2):
yield CircuitInstruction(rot, [qubits[j], qubits[j + 1]])
yield CircuitInstruction(
rot, [qubits[norb + j], qubits[norb + j + 1]]
)
yield CircuitInstruction(rot, [qubits[j + 1], qubits[j + 2]])
yield CircuitInstruction(
rot, [qubits[norb + j + 1], qubits[norb + j + 2]]
)
def trotter_step(
qubits: Sequence[Qubit],
time_step: float,
one_body_evolution: np.ndarray,
h2e: np.ndarray,
impurity_index: int,
norb: int,
):
"""A Trotter step."""
# Assume the two-body interaction is just the on-site interaction of the impurity
onsite = h2e[
impurity_index, impurity_index, impurity_index, impurity_index
]
# Two-body evolution for half the time
yield CircuitInstruction(
CPhaseGate(-0.5 * time_step * onsite),
[qubits[impurity_index], qubits[norb + impurity_index]],
)
# One-body evolution for the full time
yield CircuitInstruction(
ffsim.qiskit.OrbitalRotationJW(norb, one_body_evolution), qubits
)
# Two-body evolution for half the time
yield CircuitInstruction(
CPhaseGate(-0.5 * time_step * onsite),
[qubits[impurity_index], qubits[norb + impurity_index]],
)
# Time step
time_step = 0.2
# Number of Krylov basis states
krylov_dim = 8
# Initialize circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# Generate initial state
for instruction in prepare_initial_state(qubits, norb=norb, nocc=norb // 2):
circuit.append(instruction)
circuit.measure_all()
# Create list of circuits, starting with the initial state circuit
circuits = [circuit.copy()]
# Add time evolution circuits to the list
one_body_evolution = scipy.linalg.expm(-1j * time_step * h1e_momentum)
for i in range(krylov_dim - 1):
# Remove measurements
circuit.remove_final_measurements()
# Append another Trotter step
for instruction in trotter_step(
qubits,
time_step,
one_body_evolution,
h2e_momentum,
impurity_index,
norb,
):
circuit.append(instruction)
# Measure qubits
circuit.measure_all()
# Add a copy of the circuit to the list
circuits.append(circuit.copy())
ต่อไป เราสร้าง Circuit เพื่อสร้างสถานะฐาน Krylov สำหรับแต่ละชนิดสปิน สถานะเริ่มต้น ถูกกำหนดโดยการซ้อนทับของการกระตุ้นทั้งหมดที่เป็นไปได้ของอิเล็กตรอนสามตัวที่ใกล้กับระดับ Fermi มากที่สุดไปยังโหมดว่างที่ใกล้ที่สุด 4 โหมด โดยเริ่มจากสถานะ และทำได้โดยการใช้ XXPlusYYGate เจ็ดตัว สถานะที่วิวัฒนาการตามเวลาถูกสร้างขึ้นโดยการใช้ขั้นตอน Trotter อันดับสองซ้ำ
สำหรับคำอธิบายโดยละเอียดเพิ่มเติมเกี่ยวกับโมเดลนี้และวิธีการออกแบบ Circuit โปรดดู "Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization"
circuits[0].draw("mpl", scale=0.4, fold=-1)
circuits[-1].draw("mpl", scale=0.4, fold=-1)
ขั้นตอนที่ 2: ปรับแต่งปัญหาสำหรับการรันแบบควอนตัม
ต่อไป เราจะ optimize วงจรสำหรับ hardware เป้าหมาย สำหรับตอนนี้ เราจะสร้าง backend ทั่วไปที่มีจำนวน Qubit ที่กำหนดและชุด gate ที่ time evolution circuit สามารถ decompose ได้โดยธรรมชาติ
from qiskit.providers.fake_provider import GenericBackendV2
backend = GenericBackendV2(
2 * norb, basis_gates=["cp", "xx_plus_yy", "p", "x"]
)
ตอนนี้เราใช้ Qiskit เพื่อ transpile Circuit สำหรับ backend เป้าหมาย
from qiskit.transpiler import generate_preset_pass_manager
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend
)
isa_circuits = pass_manager.run(circuits)
ขั้นตอนที่ 3: รันด้วย Qiskit primitives
หลังจาก optimize วงจรสำหรับการรันบน hardware แล้ว เราก็พร้อมรันบน hardware จริงและเก็บตัวอย่างสำหรับการประมาณ ground state energy แล้ว หลังจากใช้ Sampler primitive เพื่อ sample bitstring จากแต่ละวงจร เราจะรวมผลลัพธ์ทั้งหมดเข้าด้วยกันเป็น counts dictionary เดียว และ plot 20 bitstring ที่ถูก sample บ่อยที่สุด
from qiskit.visualization import plot_histogram
from qiskit.primitives import StatevectorSampler
# Sample from the circuits
sampler = StatevectorSampler()
job = sampler.run(isa_circuits, shots=500)
from qiskit.primitives import BitArray
# Combine the shots from the individual Trotter circuits
bit_array = BitArray.concatenate_shots(
[result.data.meas for result in job.result()]
)
plot_histogram(bit_array.get_counts(), number_to_keep=20)
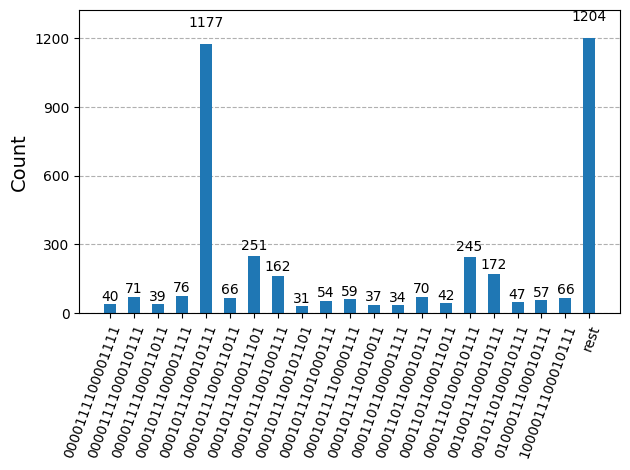
ขั้นตอนที่ 4: Post-process และคืนผลลัพธ์เป็น format ที่ต้องการ
ตอนนี้เราจะรัน SQD algorithm โดยใช้ฟังก์ชัน diagonalize_fermionic_hamiltonian ดู API documentation สำหรับคำอธิบาย argument ของฟังก์ชันนี้
from qiskit_addon_sqd.fermion import (
SCIResult,
diagonalize_fermionic_hamiltonian,
)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
rng = np.random.default_rng(24)
result = diagonalize_fermionic_hamiltonian(
h1e_momentum,
h2e_momentum,
bit_array,
samples_per_batch=100,
norb=norb,
nelec=nelec,
num_batches=3,
max_iterations=5,
symmetrize_spin=True,
callback=callback,
seed=rng,
)
Iteration 1
Subsample 0
Energy: -13.4222953188441
Subspace dimension: 529
Subsample 1
Energy: -13.42237556285828
Subspace dimension: 784
Subsample 2
Energy: -13.422045397387413
Subspace dimension: 529
Iteration 2
Subsample 0
Energy: -13.422379583305478
Subspace dimension: 900
Subsample 1
Energy: -13.422376197704326
Subspace dimension: 841
Subsample 2
Energy: -13.422421162849295
Subspace dimension: 1089
Iteration 3
Subsample 0
Energy: -13.422421164670345
Subspace dimension: 1156
Subsample 1
Energy: -13.422421492737689
Subspace dimension: 1156
Subsample 2
Energy: -13.422421205869572
Subspace dimension: 1156
Iteration 4
Subsample 0
Energy: -13.422421494558726
Subspace dimension: 1225
Subsample 1
Energy: -13.422421492737689
Subspace dimension: 1156
Subsample 2
Energy: -13.422421492737689
Subspace dimension: 1156
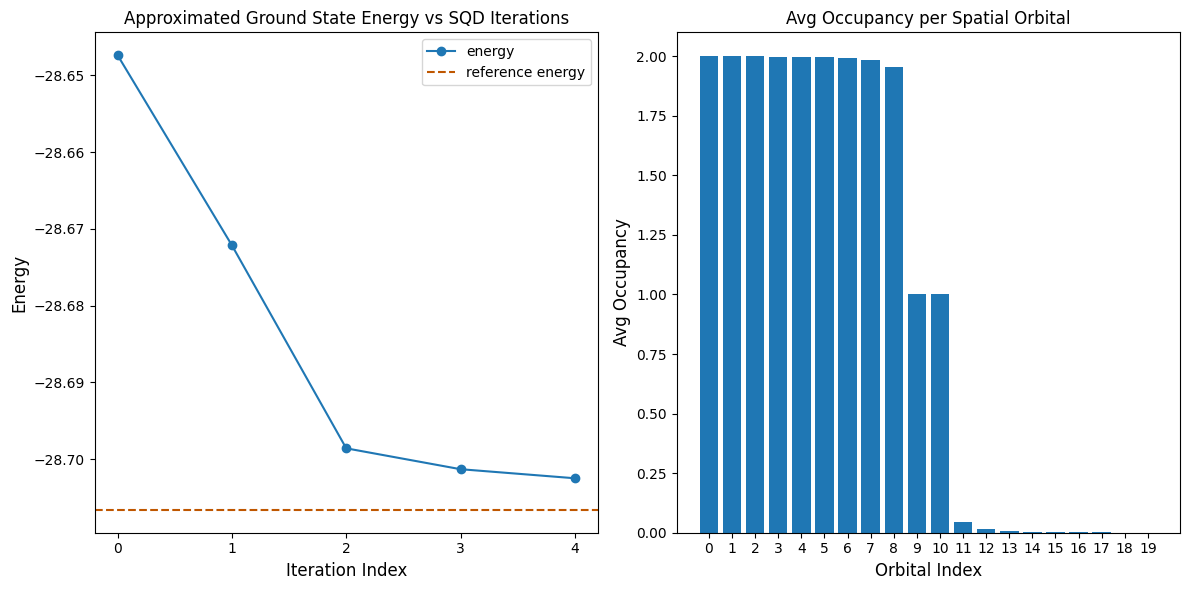
code cell ต่อไปนี้ plot ผลลัพธ์ plot แรกแสดง energy ที่คำนวณได้เป็นฟังก์ชันของจำนวน configuration recovery iteration และ plot ที่สองแสดง average occupancy ของแต่ละ spatial orbital หลังจาก iteration สุดท้าย เนื่องจากปัญหานี้มีขนาดเล็กมาก iteration แรกจึงนำเราไปใกล้ energy ที่แน่นอนมากแล้ว (สังเกตสเกลของแกน y)
import matplotlib.pyplot as plt
min_es = [
min(result, key=lambda res: res.energy).energy
for result in result_history
]
min_id, min_e = min(enumerate(min_es), key=lambda x: x[1])
# Data for energies plot
x1 = range(len(result_history))
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, min_es, label="energy", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].axhline(
y=reference_energy,
color="#BF5700",
linestyle="--",
label="reference energy",
)
axs[0].set_title("Approximated Ground State Energy vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Reference energy: {reference_energy:.5f}")
print(f"SQD energy: {min_e:.5f}")
print(f"Absolute error: {abs(min_e - reference_energy):.5f}")
plt.tight_layout()
plt.show()
Reference energy: -13.42249
SQD energy: -13.42242
Absolute error: 0.00007
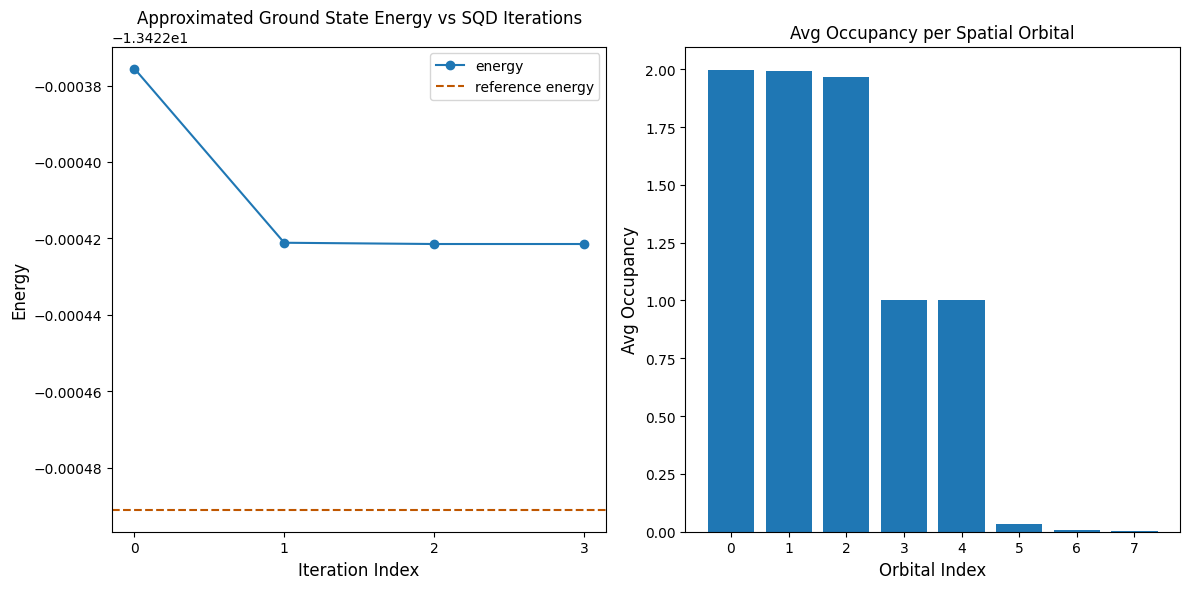
ตรวจสอบ energy
energy ที่ SQD คืนมารับประกันว่าเป็น upper bound ของ true ground state energy จริง ค่า energy สามารถตรวจสอบได้เพราะ SQD ยังคืน coefficient ของ state vector ที่ประมาณ ground state ด้วย เราสามารถคำนวณ energy จาก state vector โดยใช้ reduced density matrix หนึ่งอนุภาคและสองอนุภาค ดังที่แสดงใน code cell ต่อไปนี้
rdm1 = result.sci_state.rdm(rank=1, spin_summed=True)
rdm2 = result.sci_state.rdm(rank=2, spin_summed=True)
energy = np.sum(h1e_momentum * rdm1) + 0.5 * np.sum(h2e_momentum * rdm2)
print(f"Recomputed energy: {energy:.5f}")
Recomputed energy: -13.42242
ตัวอย่างขนาดใหญ่บน hardware จริง
ตอนนี้เราจะรันตัวอย่างที่ใหญ่ขึ้นบน QPU จริง สำหรับ reference energy เราใช้ผลลัพธ์จากการคำนวณ DMRG ที่ทำแยกต่างหาก
from qiskit_ibm_runtime import SamplerV2 as Sampler
from qiskit_ibm_runtime import QiskitRuntimeService
# Model parameters
norb = 20
nelec = (norb // 2, norb // 2)
n_bath = norb - 1
hybridization = 1.0
hopping = 1.0
onsite = 10.0
chemical_potential = -0.5 * onsite
# Generate Hamiltonian and orbital rotation
h1e, h2e = siam_hamiltonian(
norb=norb,
hopping=hopping,
onsite=onsite,
hybridization=hybridization,
chemical_potential=chemical_potential,
)
orbital_rotation = momentum_basis(norb)
h1e_momentum, h2e_momentum = rotated(h1e, h2e, orbital_rotation.T.conj())
impurity_index = n_bath // 2
# Set reference energy to DMRG value computed separately
reference_energy = -28.70659686
# Algorithm parameters
time_step = 0.2
krylov_dim = 8
# Construct circuits
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
for instruction in prepare_initial_state(qubits, norb=norb, nocc=norb // 2):
circuit.append(instruction)
circuit.measure_all()
circuits = [circuit.copy()]
one_body_evolution = scipy.linalg.expm(-1j * time_step * h1e_momentum)
for i in range(krylov_dim - 1):
circuit.remove_final_measurements()
for instruction in trotter_step(
qubits,
time_step,
one_body_evolution,
h2e_momentum,
impurity_index,
norb,
):
circuit.append(instruction)
circuit.measure_all()
circuits.append(circuit.copy())
# Initialize hardware backend
service = QiskitRuntimeService()
backend = service.least_busy(
operational=True, simulator=False, min_num_qubits=127
)
print(f"Using backend {backend.name}")
# Transpile to backend
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend
)
isa_circuits = pass_manager.run(circuits)
# Sample from the circuits
sampler = Sampler(backend)
sampler.options.environment.job_tags = ["TUT_SKQD"]
job = sampler.run(isa_circuits, shots=500)
# Combine the shots from the individual Trotter circuits
bit_array = BitArray.concatenate_shots(
[result.data.meas for result in job.result()]
)
# Run configuration recovery and diagonalization
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
rng = np.random.default_rng(24)
result = diagonalize_fermionic_hamiltonian(
h1e_momentum,
h2e_momentum,
bit_array,
samples_per_batch=100,
norb=norb,
nelec=nelec,
num_batches=3,
max_iterations=5,
symmetrize_spin=True,
callback=callback,
seed=rng,
)
# Plot results
min_es = [
min(result, key=lambda res: res.energy).energy
for result in result_history
]
min_id, min_e = min(enumerate(min_es), key=lambda x: x[1])
x1 = range(len(result_history))
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
axs[0].plot(x1, min_es, label="energy", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].axhline(
y=reference_energy,
color="#BF5700",
linestyle="--",
label="reference energy",
)
axs[0].set_title("Approximated Ground State Energy vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy", fontdict={"fontsize": 12})
axs[0].legend()
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Reference energy: {reference_energy:.5f}")
print(f"SQD energy: {min_e:.5f}")
print(f"Absolute error: {abs(min_e - reference_energy):.5f}")
plt.tight_layout()
plt.show()
Using backend ibm_boston
Iteration 1
Subsample 0
Energy: -28.63965951544449
Subspace dimension: 9801
Subsample 1
Energy: -28.625588929202006
Subspace dimension: 9409
Subsample 2
Energy: -28.647371834135498
Subspace dimension: 8281
Iteration 2
Subsample 0
Energy: -28.67213260849567
Subspace dimension: 29584
Subsample 1
Energy: -28.670340686158816
Subspace dimension: 27225
Subsample 2
Energy: -28.669976379525988
Subspace dimension: 31329
Iteration 3
Subsample 0
Energy: -28.68622875601382
Subspace dimension: 36100
Subsample 1
Energy: -28.698569623143126
Subspace dimension: 34225
Subsample 2
Energy: -28.694848533971882
Subspace dimension: 33856
Iteration 4
Subsample 0
Energy: -28.69883392844593
Subspace dimension: 42025
Subsample 1
Energy: -28.701289495200996
Subspace dimension: 38025
Subsample 2
Energy: -28.699319594978245
Subspace dimension: 45369
Iteration 5
Subsample 0
Energy: -28.701936886834154
Subspace dimension: 51076
Subsample 1
Energy: -28.702468711812013
Subspace dimension: 53824
Subsample 2
Energy: -28.702298147575938
Subspace dimension: 52900
Reference energy: -28.70660
SQD energy: -28.70247
Absolute error: 0.00413
ขั้นตอนถัดไป
หากคุณสนใจเนื้อหานี้ คุณอาจสนใจเนื้อหาต่อไปนี้:
- Sample-based quantum diagonalization of a chemistry Hamiltonian - บทแนะนำที่เกี่ยวข้องซึ่งใช้ heuristic variational ansatz แทน Trotter circuit
- Krylov quantum diagonalization of lattice Hamiltonians - บทแนะนำเกี่ยวกับวิธี KQD
- SQD addon API documentation - เอกสารอ้างอิงสำหรับฟังก์ชัน
diagonalize_fermionic_hamiltonian - Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization - บทความวิจัยที่บทแนะนำนี้อ้างอิง