การทำ Diagonalization ควอนตัมแบบอิงตัวอย่างของ Hamiltonian ทางเคมี
ประมาณการการใช้งาน: น้อยกว่าหนึ่งนาทีบนโปรเซสเซอร์ Heron r2 (หมายเหตุ: นี่เป็นการประมาณการเท่านั้น รันไทม์จริงอาจแตกต่างกัน)
ผลลัพธ์การเรียนรู้
หลังจากผ่านบทแนะนำนี้แล้ว ผู้ใช้ควรเข้าใจ:
- วิธีใช้ SQD Qiskit addon เพื่อประมาณพลังงาน ground state ของระบบโมเลกุลโดยใช้ bitstring ที่สุ่มมาจาก quantum processing unit (QPU)
- วิธีใช้ ffsim เพื่อสร้าง Circuit local unitary cluster Jastrow (LUCJ) สำหรับการจำลองเคมีควอนตัม
ข้อกำหนดเบื้องต้น
เราแนะนำให้ผู้ใช้ทำความคุ้นเคยกับหัวข้อต่อไปนี้ก่อนผ่านบทแนะนำนี้:
- เคมีควอนตัมและ second quantization
- การใช้ Sampler primitive เพื่อสุ่มตัวอย่างจาก Circuit ควอนตัม
พื้นฐาน
ในบทแนะนำนี้ เราจะแสดงวิธีการ post-process ตัวอย่างควอนตัมที่มีสัญญาณรบกวนเพื่อประมาณ ground state ของโมเลกุลไนโตรเจน ที่ความยาวพันธะสมดุล โดยใช้ SQD Qiskit addon เพื่อใช้งานอัลกอริทึม sample-based quantum diagonalization (SQD) ดูรายละเอียดเพิ่มเติมเกี่ยวกับซอฟต์แวร์ได้ที่เอกสารประกอบ รวมถึงตัวอย่างง่าย ๆ สำหรับเริ่มต้น
บทแนะนำนี้แนะนำสำหรับผู้ใช้ที่คุ้นเคยกับเคมีควอนตัม โดยเฉพาะความคุ้นเคยกับการหาพลังงาน ground state ของโมเลกุล สำหรับคำอธิบายเชิงลึกเกี่ยวกับ workflow ให้ดูที่ quantum diagonalization algorithm course
SQD เป็นเทคนิคสำหรับหา eigenvalue และ eigenvector ของ operator ควอนตัม เช่น Hamiltonian ของระบบควอนตัม โดยใช้การประมวลผลควอนตัมและคลาสสิกแบบกระจายร่วมกัน การประมวลผลคลาสสิกแบบกระจายถูกใช้เพื่อประมวลผลตัวอย่างที่ได้จากโปรเซสเซอร์ควอนตัม และเพื่อ project และ diagonalize Hamiltonian เป้าหมายในซับสเปซที่ตัวอย่างเหล่านั้น span ขั้นตอนการทำงานของ SQD มีดังนี้:
- เลือก circuit ansatz และนำไปใช้กับคอมพิวเตอร์ควอนตัมบน reference state (ในกรณีนี้คือสถานะHartree-Fock)
- สุ่ม bitstring จากสถานะควอนตัมที่ได้
- รันกระบวนการ self-consistent configuration recovery บน bitstring เพื่อหาค่าประมาณ ground state
SQD เป็นที่ทราบกันว่าทำงานได้ดีเมื่อ eigenstate เป้าหมายมีความ sparse นั่นคือ wave function ถูก support ในชุดของ basis state ที่มีขนาดไม่เพิ่มขึ้นแบบเอกซ์โปเนนเชียลตามขนาดของปัญหา
เคมีควอนตัม
Hamiltonian ของระบบโมเลกุลสามารถเขียนได้เป็น
โดยที่ และ คือจำนวนเชิงซ้อนที่เรียกว่า molecular integral ซึ่งสามารถคำนวณได้จากข้อมูลจำเพาะของโมเลกุลโดยใช้โปรแกรมคอมพิวเตอร์ ในบทแนะนำนี้ เราคำนวณ integral โดยใช้แพ็กเกจซอฟต์แวร์ PySCF
สำหรับรายละเอียดเกี่ยวกับวิธีการได้มาซึ่ง Hamiltonian ของโมเลกุล ให้ดูในตำราเรียนเคมีควอนตัม (เช่น Modern Quantum Chemistry โดย Szabo และ Ostlund) สำหรับคำอธิบายระดับสูงเกี่ยวกับการ map ปัญหาเคมีควอนตัมไปยังคอมพิวเตอร์ควอนตัม ดูการบรรยาย Mapping Problems to Qubits จาก Qiskit Global Summer School 2024
Local unitary cluster Jastrow (LUCJ) ansatz
SQD ต้องการ circuit ansatz ควอนตัมเพื่อดึงตัวอย่าง ในบทแนะนำนี้ เราจะใช้ local unitary cluster Jastrow (LUCJ) ansatz เนื่องจากการผสมผสานระหว่างแรงจูงใจทางฟิสิกส์และความเป็นมิตรกับฮาร์ดแวร์ และจะใช้ ffsim เพื่อสร้าง Circuit ansatz
LUCJ ansatz ปรับตัวให้เข้ากับ QPU ที่มีการเชื่อมต่อ Qubit จำกัด spin orbital จะถูก map ไปยัง Qubit เพื่อให้ ansatz ไม่ต้องใช้การ routing ด้วย SWAP gate ฮาร์ดแวร์ IBM® มี topology ของ Qubit แบบ heavy-hex lattice ซึ่งในกรณีนี้เราสามารถใช้รูปแบบ "zig-zag" ตามที่แสดงด้านล่าง ในรูปแบบนี้ orbital ที่มี spin เดียวกันจะถูก map ไปยัง Qubit ที่มี line topology (วงกลมสีแดงและสีน้ำเงิน) และการเชื่อมต่อระหว่าง orbital ที่มี spin ต่างกันมีอยู่ทุก spatial orbital ที่ 4 โดยการเชื่อมต่อจะถูกอำนวยความสะดวกโดย ancilla Qubit (วงกลมสีม่วง)
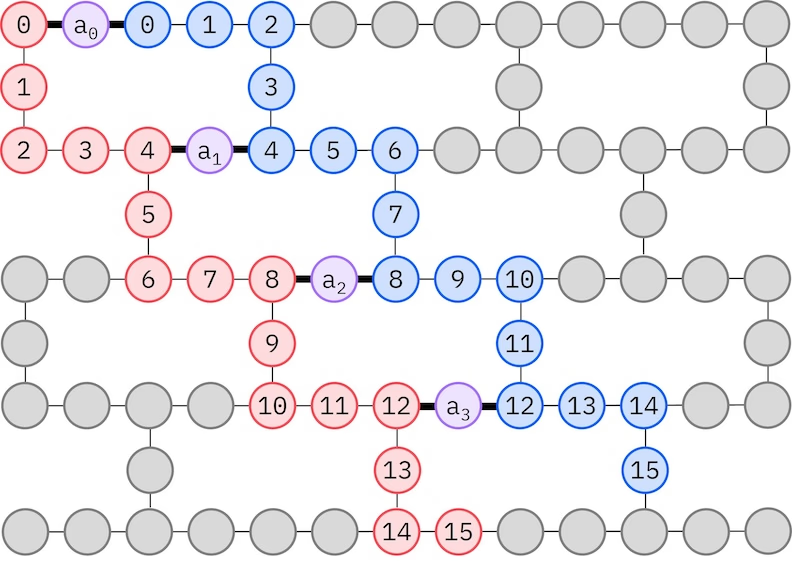
Self-consistent configuration recovery
กระบวนการ self-consistent configuration recovery ถูกออกแบบมาเพื่อดึงสัญญาณให้ได้มากที่สุดจากตัวอย่างควอนตัมที่มีสัญญาณรบกวน เนื่องจาก Hamiltonian ของโมเลกุลอนุรักษ์จำนวนอนุภาคและ spin Z จึงสมเหตุสมผลที่จะเลือก circuit ansatz ที่อนุรักษ์สมมาตรเหล่านี้ด้วย เมื่อนำไปใช้กับสถานะ Hartree-Fock สถานะที่ได้จะมีจำนวนอนุภาคและ spin Z คงที่ในการตั้งค่าที่ไม่มีสัญญาณรบกวน ดังนั้น ครึ่ง spin- และ spin- ของ bitstring ใด ๆ ที่สุ่มจากสถานะนี้ควรมีHamming weight เดียวกับสถานะ Hartree-Fock เนื่องจากมีสัญญาณรบกวนในโปรเซสเซอร์ควอนตัมปัจจุบัน bitstring บางอันที่วัดได้จะละเมิดคุณสมบัตินี้ รูปแบบง่าย ๆ ของ postselection จะทิ้ง bitstring เหล่านี้ แต่นั่นสิ้นเปลือง เพราะ bitstring เหล่านั้นอาจยังมีสัญญาณอยู่บ้าง กระบวนการ self-consistent recovery พยายามกู้คืนสัญญาณบางส่วนนั้นใน post-processing กระบวนการนี้เป็นแบบวนซ้ำและต้องการเป็น input ค่าประมาณของค่าครอบครองเฉลี่ยของแต่ละ orbital ใน ground state ซึ่งคำนวณจากตัวอย่างดิบก่อน กระบวนการจะรันในลูป และแต่ละรอบมีขั้นตอนดังนี้:
- สำหรับ bitstring แต่ละอันที่ละเมิดสมมาตรที่ระบุ ให้พลิก bit ด้วยกระบวนการแบบความน่าจะเป็นที่ออกแบบมาเพื่อให้ bitstring ใกล้เคียงกับค่าประมาณปัจจุบันของค่าครอบครอง orbital เฉลี่ยมากขึ้น เพื่อให้ได้ bitstring ใหม่
- รวบรวม bitstring เก่าและใหม่ทั้งหมดที่ตอบสนองสมมาตร และสุ่มตัวอย่างย่อยของขนาดคงที่ที่กำหนดไว้ล่วงหน้า
- สำหรับชุดย่อยของ bitstring แต่ละชุด ให้ project Hamiltonian ลงในซับสเปซที่ถูก span โดย basis vector ที่สอดคล้องกัน (ดูส่วนก่อนหน้า สำหรับคำอธิบายของ basis vector เหล่านี้) และคำนวณค่าประมาณ ground state ของ Hamiltonian ที่ถูก project บนคอมพิวเตอร์คลาสสิก
- อัปเดตค่าประมาณของค่าครอบครอง orbital เฉลี่ยด้วยค่าประมาณ ground state ที่มีพลังงานต่ำที่สุด
แผนภาพขั้นตอนการทำงาน SQD
ขั้นตอนการทำงาน SQD แสดงในแผนภาพต่อไปนี้:
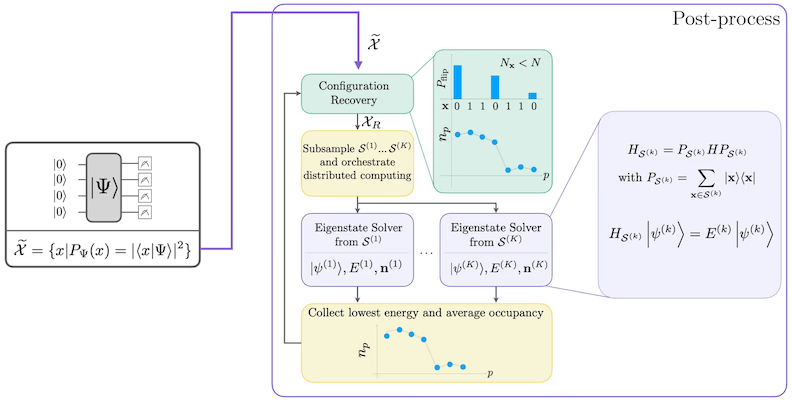
ข้อกำหนด
ก่อนเริ่มบทแนะนำนี้ ให้แน่ใจว่าได้ติดตั้งสิ่งต่อไปนี้แล้ว:
- Qiskit SDK v1.0 หรือใหม่กว่า พร้อมรองรับการแสดงผล
- Qiskit Runtime v0.22 หรือใหม่กว่า (
pip install qiskit-ibm-runtime) - SQD Qiskit addon v0.11 หรือใหม่กว่า (
pip install qiskit-addon-sqd) - ffsim v0.0.75 หรือใหม่กว่า (
pip install ffsim)
การตั้งค่า
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime
import math
import ffsim
import matplotlib.pyplot as plt
import numpy as np
import pyscf
import pyscf.cc
import pyscf.mcscf
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.primitives import StatevectorSampler
from qiskit.providers.fake_provider import GenericBackendV2
from qiskit_ibm_runtime import QiskitRuntimeService
from qiskit_ibm_runtime import SamplerV2 as Sampler
ตัวอย่างจำลองขนาดเล็ก
ในบทแนะนำนี้ เราจะหาค่าประมาณของ ground state ของโมเลกุลไนโตรเจนใกล้ระยะพันธะสมดุล เราใช้ STO-6G basis set ขนาดเล็กก่อน เพื่อให้สามารถจำลองการทดลองและตรวจสอบว่าทำงานได้ถูกต้อง
ขั้นตอนที่ 1: Map input คลาสสิกไปยังปัญหาควอนตัม
ก่อนอื่น เราระบุโมเลกุลและคุณสมบัติของมัน
# Specify molecule properties
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="sto-6g",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
norb = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
n_alpha = (n_electrons + mol.spin) // 2
n_beta = (n_electrons - mol.spin) // 2
nelec = (n_alpha, n_beta)
cas = pyscf.mcscf.CASCI(scf, norb, nelec)
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), norb)
# Compute exact energy using FCI
reference_energy = cas.run().e_tot
print(f"norb = {norb}")
print(f"nelec = {nelec}")
converged SCF energy = -108.464957764796
CASCI E = -108.595987350986 E(CI) = -32.4115475088426 S^2 = 0.0000000
norb = 8
nelec = (5, 5)
ก่อนสร้าง Circuit LUCJ ansatz เราทำการคำนวณ CCSD ในเซลล์โค้ดต่อไปนี้ก่อน amplitude และ จากการคำนวณนี้จะถูกใช้เพื่อ initialize พารามิเตอร์ของ ansatz
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -108.5933309085008 E_corr = -0.1283731437052354
ตอนนี้ เราใช้ ffsim เพื่อสร้าง Circuit ansatz เนื่องจากโมเลกุลของเรามีสถานะ Hartree-Fock แบบ closed-shell เราจึงใช้ตัวแปรที่สมดุลด้าน spin ของ UCJ ansatz ได้แก่ UCJOpSpinBalanced เราตั้ง optimize=True ในเมธอด from_t_amplitudes เพื่อเปิดใช้การ double-factorization แบบ "compressed" ของ amplitude (ดู The local unitary cluster Jastrow (LUCJ) ansatz ในเอกสาร ffsim สำหรับรายละเอียด)
เนื่องจาก LUCJ ansatz ปรับตัวให้เข้ากับการเชื่อมต่อที่มีของ QPU เราจึงต้อง initialize QPU backend ก่อนสร้าง ansatz สำหรับตอนนี้ เราจะสร้าง backend ทั่วไปที่มี heavy-hex coupling map และชุด gate ที่ LUCJ ansatz สามารถ decompose ได้ตามธรรมชาติ จากนั้นเราจะใช้ ffsim.qiskit.generate_lucj_pass_manager เพื่อสร้าง pass manager ที่เชี่ยวชาญสำหรับการ transpile LUCJ ansatz ไปยัง backend ที่กำหนดตามรูปแบบ "zig-zag" ที่อธิบายในส่วนพื้นฐานเกี่ยวกับ LUCJ ansatz ฟังก์ชันนี้ใช้ scoring heuristic เพื่อลดข้อผิดพลาดที่เกี่ยวข้องกับ layout ที่เลือก ซึ่งสำคัญหากฮาร์ดแวร์ของคุณเป็น QPU จริงหรือ simulator ที่มี noise model นอกจากการคืน pass manager แล้ว ฟังก์ชันนี้ยังคืน alpha-beta coupling pair ที่สามารถใช้บนฮาร์ดแวร์ได้ด้วย หากไม่สามารถใช้ทุก pair ได้ จะมีการแจ้งเตือน
import warnings
from qiskit.transpiler import CouplingMap
warnings.formatwarning = lambda msg, *args, **kwargs: f"Warning: {msg}\n"
# Set ansatz properties
n_reps = 1
pairs_aa = [(p, p + 1) for p in range(norb - 1)]
# Let generate_lucj_pass_manager determine the alpha-beta interactions
pairs_ab = None
# Initialize backend
coupling_map = CouplingMap.from_heavy_hex(3)
backend = GenericBackendV2(
coupling_map.size(),
coupling_map=coupling_map,
basis_gates=["cp", "xx_plus_yy", "p", "x", "swap"],
)
# Create pass manager
pass_manager, pairs_ab = ffsim.qiskit.generate_lucj_pass_manager(
backend=backend,
norb=norb,
connectivity="heavy-hex",
interaction_pairs=(pairs_aa, pairs_ab),
optimization_level=3,
)
# Create the LUCJ ansatz operator
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(pairs_aa, pairs_ab),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell
# from running too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
# create an empty quantum circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it
# to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(norb, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
ขั้นตอนที่ 2: ปรับแต่งสำหรับการรันบนฮาร์ดแวร์ควอนตัม
ต่อไป เราปรับแต่ง Circuit สำหรับฮาร์ดแวร์เป้าหมาย โดยปกติแล้วขั้นตอนนี้เกี่ยวข้องกับการ initialize hardware backend และ pass manager สำหรับ backend นั้น แต่เนื่องจาก LUCJ ansatz ปรับตัวให้เข้ากับการเชื่อมต่อของฮาร์ดแวร์ เราได้ดำเนินการเหล่านั้นในขั้นตอนก่อนหน้าแล้ว สิ่งที่เหลืออยู่คือการรัน pass manager บน Circuit เพื่อ transpile เป็น ISA circuit ที่สามารถรันบน QPU ได้โดยตรง
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts: {isa_circuit.count_ops()}")
Gate counts: OrderedDict({'xx_plus_yy': 86, 'p': 16, 'measure': 16, 'cp': 15, 'x': 10, 'swap': 2, 'barrier': 1})
ขั้นตอนที่ 3: รันด้วย Qiskit primitives
หลังจากปรับแต่ง Circuit ให้พร้อมสำหรับการรันบนฮาร์ดแวร์แล้ว เราก็พร้อมรันบนฮาร์ดแวร์จริงและเก็บตัวอย่างสำหรับการประมาณพลังงาน ground state เนื่องจากมีเพียง Circuit เดียว เราจะใช้ Job execution mode ของ Qiskit Runtime เพื่อรัน Circuit
rng = np.random.default_rng()
sampler = StatevectorSampler(seed=rng)
job = sampler.run([isa_circuit], shots=100_000)
Warning: Trying to add QuantumRegister to a QuantumCircuit having a layout
primitive_result = job.result()
pub_result = primitive_result[0]
ขั้นตอนที่ 4: ประมวลผลและแสดงผลลัพธ์ในรูปแบบคลาสสิก
เมตริกที่มีประโยชน์ในการประเมินคุณภาพของผลลัพธ์จาก QPU คือจำนวน configuration ที่ถูกต้องที่ได้รับ configuration ที่ถูกต้องมีจำนวนอนุภาคและ spin Z ที่ถูกต้อง ซึ่งหมายความว่าครึ่งขวาของ bitstring มี Hamming weight เท่ากับจำนวนอิเล็กตรอน spin-up และครึ่งซ้ายมี Hamming weight เท่ากับจำนวนอิเล็กตรอน spin-down เซลล์ต่อไปนี้คำนวณสัดส่วนของ configuration ที่สุ่มได้ซึ่งถูกต้อง
def is_valid_bitstring(
bitstring: str, norb: int, nelec: tuple[int, int]
) -> bool:
n_alpha, n_beta = nelec
return (
len(bitstring) == 2 * norb
and bitstring[norb:].count("1") == n_alpha
and bitstring[:norb].count("1") == n_beta
)
bit_array = pub_result.data.meas
num_valid = sum(
is_valid_bitstring(b, norb, nelec) for b in bit_array.get_bitstrings()
)
valid_fraction = num_valid / bit_array.num_shots
print(f"Fraction of sampled configurations that are valid: {valid_fraction}")
Fraction of sampled configurations that are valid: 1.0
bitstring ทั้งหมดถูกต้องเนื่องจากเราสุ่มตัวอย่าง Circuit บน simulator ที่ไม่มี noise เมื่อรันบน QPU ที่มี noise สัดส่วนจะน้อยกว่าหนึ่ง แต่หวังว่าจะมากกว่าสัดส่วนที่คาดหากสุ่ม bitstring แบบ uniform ซึ่งคำนวณได้ในเซลล์ต่อไปนี้
expected_fraction_random = (
math.comb(norb, n_alpha) * math.comb(norb, n_beta) / 2 ** (2 * norb)
)
print(
f"Expected fraction of valid configurations from uniformly random bitstrings: "
f"{expected_fraction_random}"
)
Expected fraction of valid configurations from uniformly random bitstrings: 0.0478515625
ตอนนี้เราจะประมาณพลังงาน ground state ของ Hamiltonian โดยใช้ฟังก์ชัน diagonalize_fermionic_hamiltonian ฟังก์ชันนี้ทำกระบวนการ self-consistent configuration recovery เพื่อปรับปรุงตัวอย่างควอนตัมที่มีสัญญาณรบกวนแบบวนซ้ำ เพื่อให้การประมาณพลังงานดีขึ้น เราส่ง callback function เข้าไปเพื่อบันทึกผลลัพธ์ระหว่างกลางสำหรับการวิเคราะห์ในภายหลัง ดู API documentation สำหรับคำอธิบาย argument ของ diagonalize_fermionic_hamiltonian
ที่นี่ เราใช้ argument initial_occupancies ของ diagonalize_fermionic_hamiltonian เพื่อระบุ Hartree-Fock configuration เป็นค่าเริ่มต้นสำหรับค่าครอบครอง orbital ใน ground state วิธีนี้เหมาะสมสำหรับระบบที่ ground state มี support มากบน Hartree-Fock configuration แต่อาจไม่เหมาะในสถานการณ์อื่น แม้ว่าวิธีการคำนวณขั้นสูงกว่าอาจให้ค่าเริ่มต้นที่ดีกว่าในกรณีเหล่านั้น การระบุ initial_occupancies ยังช่วยให้ configuration recovery สามารถรันได้แม้ว่าจะไม่มี configuration ที่ถูกต้องถูกสุ่มมาเลย ซึ่งอาจเกิดขึ้นเมื่อสุ่มตัวอย่าง Circuit ขนาดใหญ่บน QPU ที่มี noise หากไม่มี argument นี้ configuration recovery จะล้มเหลวและแสดงข้อผิดพลาดหากไม่มี configuration ที่ถูกต้องถูกส่งเข้ามา
from functools import partial
from qiskit_addon_sqd.fermion import (
SCIResult,
diagonalize_fermionic_hamiltonian,
solve_sci_batch,
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Use the Hartree-Fock configuration as an initial guess for the orbital occupancies
initial_occupancies = (
np.array([1] * n_alpha + [0] * (norb - n_alpha)),
np.array([1] * n_beta + [0] * (norb - n_beta)),
)
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=norb,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
initial_occupancies=initial_occupancies,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
final_energy = result.energy + nuclear_repulsion_energy
energy_error = final_energy - reference_energy
print(f"Final energy: {final_energy}")
print(f"Final energy error: {energy_error}")
Iteration 1
Subsample 0
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 1
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 2
Energy: -108.59275573641656
Subspace dimension: 900
Iteration 2
Subsample 0
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 1
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 2
Energy: -108.59275573641656
Subspace dimension: 900
Final energy: -108.59275573641656
Final energy error: 0.0032316145694579745
แสดงผลลัพธ์
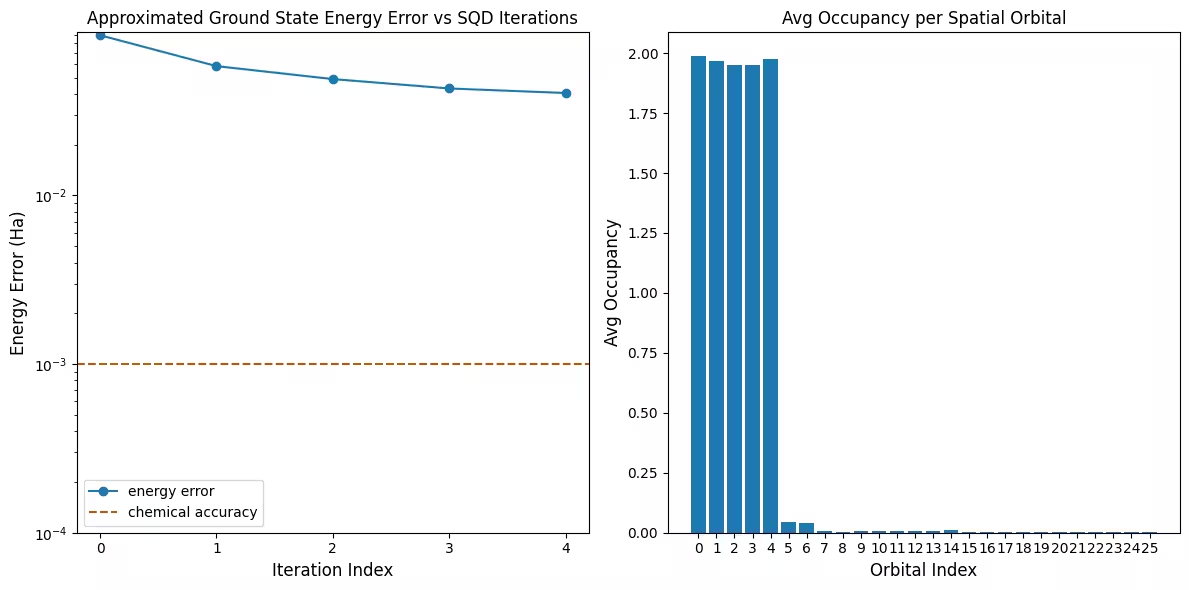
กราฟแรกแสดงให้เห็นว่าในการจำลองนี้ เราอยู่ภายใน 1 mH จากคำตอบที่แน่นอนแล้วหลังจากรอบแรก (ความแม่นยำทางเคมีโดยทั่วไปยอมรับที่ 1 kcal/mol 1.6 mH) อย่างไรก็ตาม นี่เป็นระบบขนาดเล็ก และเนื่องจากตัวอย่างไม่มี noise จึงไม่จำเป็นต้องใช้ configuration recovery บนระบบขนาดใหญ่ที่รันบน QPU ที่มี noise อาจต้องใช้หลายรอบของ configuration recovery และความแม่นยำสุดท้ายอาจแย่ลง โดยทั่วไปแล้ว พลังงานสามารถปรับปรุงได้โดยการเพิ่มจำนวนรอบของ configuration recovery หรือเพิ่มจำนวนตัวอย่างต่อ batch
กราฟที่สองแสดงค่าการครอบครองเฉลี่ยของแต่ละ spatial orbital หลังจากการวนซ้ำครั้งสุดท้าย จะเห็นได้ว่าทั้งอิเล็กตรอน spin-up และ spin-down ครอบครอง 5 orbital แรกด้วยความน่าจะเป็นสูงในคำตอบของเรา
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - reference_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()

ตัวอย่างฮาร์ดแวร์ขนาดใหญ่
ตอนนี้เราจะรันตัวอย่างที่ใหญ่ขึ้นบนฮาร์ดแวร์ควอนตัมจริง ที่นี่เราจะกำหนด active space สำหรับโมเลกุลไนโตรเจนจาก cc-pVDZ basis set
ขั้นตอนที่ 1-4
ที่นี่เราจะรวมขั้นตอนทั้งหมดเข้าด้วยกันเป็น workflow เดียวในระดับที่ใหญ่ขึ้น จากนั้นรันบนฮาร์ดแวร์ควอนตัมจริง
# ------------------------------ Step 1 ------------------------------
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="cc-pvdz",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
norb = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
n_alpha = (n_electrons + mol.spin) // 2
n_beta = (n_electrons - mol.spin) // 2
nelec = (n_alpha, n_beta)
cas = pyscf.mcscf.CASCI(scf, norb, nelec)
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), norb)
# Store reference energy from SCI calculation performed separately
reference_energy = -109.22802921665716
print(f"norb = {norb}")
print(f"nelec = {nelec}")
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
# Set ansatz properties
n_reps = 1
pairs_aa = [(p, p + 1) for p in range(norb - 1)]
# Let generate_lucj_pass_manager determine the alpha-beta interactions
pairs_ab = None
# Initialize backend
service = QiskitRuntimeService()
backend = service.least_busy(
operational=True, simulator=False, min_num_qubits=133
)
print(f"Using backend {backend.name}")
# Create pass manager
pass_manager, pairs_ab = ffsim.qiskit.generate_lucj_pass_manager(
backend=backend,
norb=norb,
connectivity="heavy-hex",
interaction_pairs=(pairs_aa, pairs_ab),
optimization_level=3,
)
# Create the LUCJ ansatz operator
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(pairs_aa, pairs_ab),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell
# from running too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
# create an empty quantum circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it
# to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(norb, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
# ------------------------------ Step 2 ------------------------------
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts: {isa_circuit.count_ops()}")
# ------------------------------ Step 3 ------------------------------
sampler = Sampler(mode=backend)
sampler.options.environment.job_tags = ["TUT_SQD"]
job = sampler.run([isa_circuit], shots=100_000)
primitive_result = job.result()
pub_result = primitive_result[0]
# ------------------------------ Step 4 ------------------------------
bit_array = pub_result.data.meas
num_valid = sum(
is_valid_bitstring(b, norb, nelec) for b in bit_array.get_bitstrings()
)
valid_fraction = num_valid / bit_array.num_shots
print(f"Fraction of sampled configurations that are valid: {valid_fraction}")
expected_fraction_random = (
math.comb(norb, n_alpha) * math.comb(norb, n_beta) / 2 ** (2 * norb)
)
print(
f"Expected fraction of valid configurations from uniformly random bitstrings: "
f"{expected_fraction_random}"
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Use the Hartree-Fock configuration as an initial guess for the
# orbital occupancies
initial_occupancies = (
np.array([1] * n_alpha + [0] * (norb - n_alpha)),
np.array([1] * n_beta + [0] * (norb - n_beta)),
)
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the
# sci_solver argument in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=norb,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
initial_occupancies=initial_occupancies,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
final_energy = result.energy + nuclear_repulsion_energy
energy_error = final_energy - reference_energy
print(f"Final energy: {final_energy}")
print(f"Final energy error: {energy_error}")
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - reference_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()
converged SCF energy = -108.929838385609
norb = 26
nelec = (5, 5)
E(CCSD) = -109.2177884185544 E_corr = -0.2879500329450045
Using backend ibm_boston
Warning: Backend cannot accommodate pairs_ab=[(0, 0), (4, 4), (8, 8), (12, 12), (16, 16), (20, 20), (24, 24)].
Removing interaction (24, 24) from the end.
Warning: Backend cannot accommodate pairs_ab=[(0, 0), (4, 4), (8, 8), (12, 12), (16, 16), (20, 20)].
Removing interaction (20, 20) from the end.
Gate counts: OrderedDict({'sx': 7039, 'rz': 6990, 'cz': 1858, 'x': 61, 'measure': 52, 'barrier': 1})
Fraction of sampled configurations that are valid: 0.02124
Expected fraction of valid configurations from uniformly random bitstrings: 9.607888706852918e-07
Iteration 1
Subsample 0
Energy: -109.13889134249762
Subspace dimension: 120409
Subsample 1
Energy: -109.11785470455858
Subspace dimension: 110889
Subsample 2
Energy: -109.13234360554011
Subspace dimension: 130321
Iteration 2
Subsample 0
Energy: -109.16392179579177
Subspace dimension: 223729
Subsample 1
Energy: -109.16281938332986
Subspace dimension: 223729
Subsample 2
Energy: -109.16955816711932
Subspace dimension: 233289
Iteration 3
Subsample 0
Energy: -109.17905772999075
Subspace dimension: 324900
Subsample 1
Energy: -109.17532445048462
Subspace dimension: 357604
Subsample 2
Energy: -109.1733168689756
Subspace dimension: 348100
Iteration 4
Subsample 0
Energy: -109.18437778820451
Subspace dimension: 474721
Subsample 1
Energy: -109.18450164209159
Subspace dimension: 476100
Subsample 2
Energy: -109.18493571190754
Subspace dimension: 487204
Iteration 5
Subsample 0
Energy: -109.18616522497996
Subspace dimension: 622521
Subsample 1
Energy: -109.18652868888333
Subspace dimension: 644809
Subsample 2
Energy: -109.18753326484406
Subspace dimension: 585225
Final energy: -109.18753326484406
Final energy error: 0.040495951813099396
ขั้นตอนถัดไป
หากคุณพบว่างานนี้น่าสนใจ คุณอาจสนใจเนื้อหาต่อไปนี้:
- Sample-based Krylov quantum diagonalization of a fermionic lattice model - บทแนะนำที่เกี่ยวข้องโดยใช้ time evolution Circuit แทน variational ansatz
- Scale SQD chemistry workflows with Dice solver - หน้าที่แสดงวิธีใช้ซอฟต์แวร์ Dice ที่มีประสิทธิภาพมากขึ้นสำหรับการ diagonalization
- SQD addon API documentation - reference สำหรับฟังก์ชัน
diagonalize_fermionic_hamiltonian - Chemistry beyond the scale of exact diagonalization on a quantum-centric supercomputer - บทความที่ tutorial นี้อ้างอิงถึง